Drugs and Toxins that Produce Delayed Toxicities
By Matthew Stripp, MD, and Claire Seo, MD
EXECUTIVE SUMMARY
- A multitude of drugs and toxins can present with a delayed toxidrome secondary to patient factors, drug factors, and toxic metabolites.
- Ingestion of drugs or toxins that have a long latency period may lead to a delayed presentation as well as a more complicated clinical course.
- It is important to consider delayed toxicity in patients with heart, kidney, and/or hepatic dysfunction because these comorbidities may decrease the clearance and metabolism of drugs and toxins.
- Overdose of extended-release formulations of drugs may delay the onset of toxicity.
- Massive ingestions of drugs can lead to pharmacobezoar formation and alter both the release and absorption of the drug.
- For certain drugs and toxins, early provision of activated charcoal should be considered because it may decrease the absorption of the substance in the immediate phase after ingestion.
- Specific antidotes/therapies should be initiated early in suspected toxicities, such as N-acetylcysteine in acetaminophen overdose and fomepizole in toxic alcohol ingestion.
- The emergency provider should consider prolonged inpatient monitoring for certain patients depending on the suspected agent and patient symptomatology.
- The emergency provider should use the guidance of local/national poison control centers when evaluating and managing a patient presenting with suspected delayed toxicity.
“I’m just a little bit tired,” says Patient A, who intentionally overdosed on an unknown amount of multiple antidepressants. “I’m absolutely fine. Please just send me home,” says Patient B, who accidentally overdosed on an anti-inflammatory medication for chest pain 15 hours ago. Given unremarkable physical exam findings and workup, the decision is made to send these patients to the observation unit for six hours of monitoring. Patient A is asymptomatic at six hours and is discharged but then presents to the emergency department (ED) two hours later unresponsive and with a wide-complex arrhythmia. Patient B becomes altered after two consecutive seizures five hours into the observation period and ultimately is admitted to the hospital.
Drug overdose is a common chief complaint in the ED. Overdose of certain medications or ingestions of toxins should prompt the emergency physician to carefully deliberate on the appropriate next steps and disposition. Furthermore, delayed toxicity may be under-recognized, and delayed management can lead to life-threatening complications, such as arrhythmias and seizures. Managing the poisoned patient can be challenging because clinical effects often are difficult to predict in circumstances that produce toxicity. Therefore, it is imperative for emergency physicians to understand the mechanisms of delayed toxicity. A thorough understanding of various causative agents will empower the emergency provider to predict the trajectory of the clinical course of a drug or toxin, gauge when toxic effects may become apparent, and evaluate the need for gastrointestinal (GI) decontamination, extracorporeal removal, or antidote therapy.
Mechanisms of Delayed Toxicity
Factors that may contribute to delayed toxicity include xenobiotic (any foreign substance to the body)-specific characteristics, patient-specific characteristics, and the pharmaceutical formulation. Delayed gastric emptying is another factor that can produce delayed toxicity wherein manifestations of toxic effects are due to delayed peak concentration secondary to delayed absorption. In the context of this review, delayed toxicity is defined as significant clinical deterioration occurring hours to days after an initial exposure.
Xenobiotic Properties
Toxicokinetics and toxicodynamics are analogous to pharmacokinetics (dose-concentration) and pharmacodynamics (concentration-effect), respectively, when considering the toxic effects of drugs.1 The GI tract is one of the most important sites of toxin absorption. While the two major sites of drug metabolism and elimination are the kidneys and liver, drugs also can be excreted by other means, including bile, air expired by the lungs, sweat, or breast milk.2,3 Delayed toxicity also may be related to a xenobiotic’s chemical structure and its effects at the cellular level.4 A xenobiotic’s interactions with the cytochrome P450 (CYP) enzymes also can contribute to how toxicity is manifested through drug metabolism.3 Large quantities of xenobiotic exposure often lead to delayed toxic effects as metabolic pathways are overwhelmed. The formation of toxic metabolites, in addition to the parent drugs, can be a significant cause for delayed toxicity.
Patient Characteristics
Acute and chronic conditions can contribute to delayed toxicity by altering a toxin’s volume of distribution and clearance.2 Disease states that deserve special attention include heart failure, kidney and hepatic disease, and decreased gut perfusion. Decreased blood flow to any organ that metabolizes or excretes a xenobiotic or toxin can affect clearance.3 In critically ill patients, hemodynamic, metabolic, and biochemical derangements may modify pharmacokinetic parameters, leading to delayed toxic effects.5
Age is another important factor because of changes in the GI tract and renal clearance systems. Individuals at the extremes of age may be at particular risk. The GI tract undergoes a multitude of changes throughout life. For example, changes in stomach size and activity may reduce gastric emptying.6 Age-related physiologic changes in elderly patients with chronic GI diseases (e.g., gastroparesis, atrophic gastritis, peptic ulcer disease) may alter toxin absorption and increase the risk of delayed toxicity.6 Renal drug excretion naturally decreases with age as the glomerular filtration rate decreases.7 In neonates, dramatically smaller stomach volumes and immature renal clearance systems may exacerbate drug dissolution and reduce drug excretion.8
Pharmaceutical Formulation
Extended-release (ER) and delayed-release xenobiotics are designed to prolong the effects of the xenobiotic and reduce dosing frequency by controlling the rate of absorption from the GI tract.9 These preparations limit fluctuation in plasma drug concentrations to provide a more uniform therapeutic effect.7 Unfortunately, in overdose, these formulations can lead to prolonged or delayed toxic effects.
Although rare, pharmacobezoars remain a potentially lethal clinical entity, and several related fatalities have been reported over the past decades.10 Pharmacobezoars form when pharmaceutical preparations conglomerate into a mass and persist in the GI tract. They have been observed after acute massive ingestions, with both immediate-release (IR) and ER formulations, and during routine administration of medications.10-12 It is difficult to predict when bezoars will form.10-13 These concretions alter absorption through several mechanisms. First, they can cause gastric or mechanical bowel obstruction from their sheer size. Secondly, they reduce the rate of dissolution due to the lower surface to volume ratio.10,13,14 Consequently, xenobiotics are steadily released from the pharmacobezoar, delaying the onset and prolonging toxicity.
Body Packers and Stuffers
Body packing and stuffing are distinguished by the quantity of packets concealed and quality of their wrapping. Body stuffers conceal hastily wrapped drugs by swallowing them in unexpected encounters with law enforcement, making them more vulnerable to packet rupture.15 Body packers internally conceal carefully wrapped drugs for transportation purposes, mainly by ingesting large numbers of packets (up to 1 kg of drugs have been reported16). Insertion of packets into the vagina or rectum also is implemented.17 Packed drugs usually are tightly wrapped in several layers of materials (e.g., latex, aluminum foil, paraffin, fiberglass) to enhance resistance to breakdown during GI transit.16,17 The potential for delayed and life-threatening symptoms occurs when the integrity of the package is lost or damaged. The large variability in packaging can lead to varying clinical presentations. Delayed toxicity can result from the erratic absorption of a large burden of drug product.17 Asymptomatic patients typically are managed conservatively. Whole bowel irrigation and activated charcoal may be administered to accelerate intestinal transit.15,18 Surgical decontamination with laparotomy is strongly considered for patients with bowel obstruction or perforation or in those presenting with emergent toxidromes from packet rupture.15,18,19
Delayed Gastric Emptying
Delayed gastric emptying can lead to delayed peak concentration of a xenobiotic. It typically is associated with drugs that directly affect the smooth muscles of the GI tract. Two primary classes of medications with this effect are opioids and anticholinergics.14 Examples of xenobiotics with strong anticholinergic activity include antihistamines, tricyclic antidepressants, antipsychotics (especially quetiapine and olanzapine), and antimuscarinics used for bladder control, such as oxybutynin. Pylorospasm after a large overdose of salicylate tablets is another distinctive example.20
A Selection of Drugs and Toxins
The following selected xenobiotics are presented in order based on approximate general clinical significance, prevalence, and severity. See Table 1 for a summary.
Table 1. Summary of Xenobiotics that Can Present with Delayed Toxicities | |||
Xenobiotic | Toxic Metabolite(s)/Toxin | Prominent Delayed Toxic Effect(s) | Typical Onset of Delayed Toxicity* |
Bupropion |
|
|
|
Toxic alcohols
|
|
|
|
Acetaminophen |
|
|
|
Salicylate |
|
|
|
Hypoglycemics
|
|
|
|
Citalopram, escitalopram |
|
|
|
Colchicine |
|
|
|
Toxic inhalants
|
|
|
|
Chlorfenapyr |
|
|
|
Mushrooms
|
|
|
|
* Reported as within X time since exposure | |||
Bupropion
Bupropion is the antidepressant most frequently associated with overdose fatalities.21 Its chemical structure has a resemblance to “bath salts,” and it can produce a false-positive for amphetamines on urine drug screens.22 Bupropion primarily inhibits dopamine reuptake and norepinephrine to a lesser extent, and it may even increase serotoninergic neuronal firing unlike other antidepressants.23,24 There are some concerns that bupropion’s metabolites can cause toxicity.23 Therefore, the combination of an ER bupropion product and its metabolites creates concern for significantly delayed and life-threatening toxicity.
At therapeutic or chronic doses above 450 mg/day, bupropion seizure risk increases in approximately 0.1% of patients.23,25 Bupropion can lower the seizure threshold in a dose-dependent manner.25,26 It is difficult to predict which patients are at highest risk for developing seizures. Initial/sustained tachycardia and altered mental status may predict the onset of late seizures.25,27 QT prolongation (> 500 msec) and ages 13 to 18 years have been associated with an increased risk for the development of seizures in patients with bupropion ingestion.25,28 In overdose, bupropion can cause sinus tachycardia (most common finding), QRS complex prolongation (via cardiac gap junction inhibition), QT prolongation (via IKr inhibition), and cardiac arrest.23,29 Other symptoms include nonspecific GI symptoms, agitation, hallucinations, and seizures.23,28 Ventricular dysrhythmias are a serious concern but uncommon.28 For most patients, toxic effects develop within eight hours of presentation, but seizures can be significantly delayed, up to 24 hours after ingestion.23,25,27,28 In the pediatric population, symptomatology can vary by age group, with vomiting more common in younger patients and tachycardia and seizures more common in teenagers.30,31 Most patients recover without sequelae; however, deaths have been reported at ingested doses between 5.4 g and 9 g.32
After IR bupropion overdose, a minimum of approximately a 12-hour observation period often is recommended until symptom resolution.32 At the time of medical clearance, patients are recommended to be asymptomatic with normal vital signs. After an overdose of suspected ER or unknown preparation, a minimum of approximately a 24-hour observation period is recommended because of the higher risk of delayed toxicity. If patients become symptomatic, observe until symptoms are resolved. Monitoring pediatric patients at a healthcare facility is suggested if the calculated bupropion dose is unknown or greater than 10 mg/kg.30
Management is focused primarily on patient stabilization; a focus on airway, breathing, and circulation (the ABCs); and largely supportive care. Seizures may be treated using typical methods such as benzodiazepines, and possibly followed by barbiturates.33 Sodium bicarbonate by intravenous (IV) push often is considered for QRS widening. However, this may not significantly decrease QRS duration due to a suspected lack of sodium channel blocking effect from bupropion.34 The emergency provider can consider using lidocaine as a second-line agent for wide complex dysrhythmias that are refractory to sodium bicarbonate.35 Activated charcoal may have a theoretical benefit when it can be safely administered without contraindications (alterations in mental status/inability to maintain airway), especially if given within one hour of ingestion. Venous-arterial extracorporeal membrane oxygenation (VA-ECMO) has been used for massive overdoses, or in patients who present with cardiogenic shock.36,37 Intravenous lipid emulsion (ILE) often is reserved for cardiac arrest, peri-arrest, or cardiotoxicity unresponsive to other therapies and with careful consideration of risks and benefits. ILE has shown some success at the case report level for cardiovascular collapse.31 As with any overdose, management also should be coordinated and guided by local/state poison control centers and/or an available toxicologist on call.
Toxic Alcohols
While fatalities are rare, the diagnosis of toxic alcohol ingestion can be challenging and treatment delay can affect the morbidity of toxic alcohol poisonings for both accidental and intentional exposures.38,39 The term “toxic alcohol” refers to several short chain hydrocarbons other than ethanol that are not intended for ingestion. Toxic alcohols cannot be readily differentiated by their appearance because they are all colorless. Ethylene glycol (EG) is the most common exposure among toxic alcohols, and deaths have been reported with as little as 30 mL of EG ingestion.38,40 The reported minimum lethal dose of methanol in adults is estimated to be even lower at 10 mL or 0.3 g/kg to 1 g/kg of body weight.40,41
Manifestations of life-threatening toxicity in methanol and EG poisonings can present late because of the time required for toxic metabolites to accumulate. Furthermore, as a competitive substrate for alcohol dehydrogenase (ADH), co-ingested ethanol can delay the onset of toxicity by delaying the production of toxic metabolites.38,42 These toxic metabolites are mitochondrial toxins in the central nervous system (CNS) and GI tract (formaldehyde, formic acid), are inhibitors of cellular respiration (glycolic acid), and also can lead to calcium oxalate crystal formation (oxalic acid). Three common features of toxic alcohol poisonings are CNS symptoms (e.g., headaches, dizziness, seizures), nonspecific GI symptoms, and acidosis.39,43 Inebriation may be present initially, but patients with chronic alcoholism may be an exception if they have developed tolerance.43 Symptom onset in methanol poisoning can occur six to 24 hours after ingestion, with acidosis developing approximately 16 to 24 hours later.42 Visual deficits and neurologic toxicity often are permanent in methanol poisoning.38,40,43 Symptom onset in EG poisoning can occur at 12 to 24 hours after ingestion, often starting with neurologic symptoms followed by cardiopulmonary symptoms then acute kidney injury (AKI).38 Hypocalcemia may not always be present in EG poisoning.43-45
Knowledge of toxic alcohol metabolism is critical to optimizing management. (See Figure 1A-C.) The anion and osmolal gaps display a reciprocal relationship over time as metabolism progresses. (See Figure 1D.) High anion gap metabolic acidosis (HAGMA) occurs from the accumulation of potent organic acids.38,43,40 Definitive diagnosis of toxic alcohol ingestion typically requires gas chromatography. Although osmolal and anion gaps can be used as surrogate markers, neither is sensitive nor specific for diagnosing toxic alcohol poisoning.38 Furthermore, negative or small positive osmolal or anion gaps cannot be used to fully exclude toxic alcohol ingestion. However, a very high osmolal gap (> 50 mOsm/L to 70 mOsm/L) usually indicates toxic alcohol ingestion.43 Although ketosis without acidosis usually is diagnostic of isopropanol poisoning, metabolic acidosis also can be present if the patient has concomitant hypotension or coingestants.38
Figure 1. Metabolism of Toxic Alcohols |
(A) Methanol, (B) Ethylene glycol, (C) Isopropyl alcohol. In the presence of an electron receptor, toxic alcohols are oxidized sequentially by alcohol dehydrogenase (ADH) and aldehyde dehydrogenase (ALDH). Isopropanol is not further metabolized by ALDH as acetone is a ketone. Elevated osmolal gap is prominent in the early course (gray gradient bar), but as metabolism proceeds, osmolal gap declines and anion gap develops with accumulation of toxic organic acids (red gradient bar). Primary toxic metabolites contributing to metabolic acidosis are boxed. Asterisk denotes the rate-limiting step. Red arrows denote detrimental pathways. Dotted arrows denote benign pathways. (D) Reciprocal relationship between osmolal gap and anion gap over time in toxic alcohol poisoning. Coingested ethanol will contribute to the rise in osmolal gap and prolong anion gap duration. |
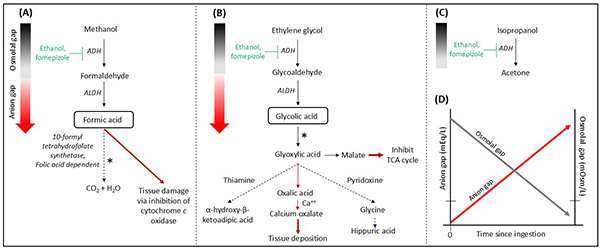 |
Adapted from: Ghannoum M, Roberts D, Bouchard J. Enhanced Elimination of Poisons. In: Yu ASL, Chertow GM, Luyckx V, et al, eds. Brenner and Rector’s The Kidney. 11th ed. Elsevier; 2020; and Wiener SW. Toxic Alcohols. In: Nelson LS, Howland MA, Lewin NA, et al, eds. Goldrank’s Toxicologic Emergencies. 11th ed. McGraw-Hill Education; 2019:1421. |
Once there is concern for toxic alcohol ingestion, important laboratory tests to consider include blood gas, lactate, serum ethanol, serum osmolality, urinalysis, and additional laboratory tests to exclude other causes of HAGMA or elevated osmolal gap (see Table 2).46 Consider serum levels of toxic alcohols if clinical suspicion is high. A “lactate gap,” which refers to a falsely elevated lactate on some point-of-care analyzers compared to laboratory analyzers, may be present from glycolic acid interfering with the lactate assay.40,43 Urine fluorescence under Wood’s lamp is considered an unreliable test to detect EG. Oxalate crystalluria may suggest EG poisoning but may not always be present.43
The goals of treatment are to prevent toxic alcohol metabolism and eliminate the alcohols and metabolites from the body. GI decontamination is not recommended because toxic alcohols are rapidly absorbed.38,43 The emergency provider should consider antidotal treatment with fomepizole, a competitive ADH antagonist, for documented history of significant exposure, serum methanol or EG concentration > 20 mg/dL, or strong suspicion of ingestion with at least two laboratory abnormalities (pH < 7.3, bicarbonate < 20 mEq/L, osmol gap > 10 mOsm/L for EG, > 20 mOsm/L for methanol).43 However, if fomepizole is unavailable, administering ethanol is recommended because ADH still has a higher affinity for ethanol than it does for toxic alcohols. Fomepizole has many advantages over ethanol, including more potent ADH inhibition, simple dosing, no need for blood monitoring, and fewer side effects.43 For adjunctive therapies, consider administering folate, thiamine, and pyridoxine because each offers the theoretical advantage of shunting metabolism toward less toxic metabolites. Consider administering sodium bicarbonate for life-threatening acidemia.47 In severe poisonings, nephrology consultation is recommended because emergent dialysis is considered a definitive therapy.
Table 2. Causes of High Anion Gap Metabolic Acidosis (HAGMA) and Elevated Osmolal Gap | ||
Causes of HAGMA | Common Causes of both HAGMA and Elevated Osmolal Gap | Causes of Elevated Osmolal Gap |
|
|
|
Acetaminophen
Acetaminophen (APAP) toxicity has consistently been reported as one of the top agents of toxic exposure.48 APAP has been a preferred analgesic since the 1970s, and is available in a variety of formulations and found in combination with many other medications (e.g., opioids, other analgesics, sedatives, decongestants, expectorants, and antihistamines).49,50 APAP has antipyretic and analgesic properties from central cyclooxygenase (COX)-2 inhibition as well as weak anti-inflammatory and antiplatelet effects.50 It is absorbed rapidly and reaches peak plasma concentrations within two hours.50 Extended-release APAP is almost entirely absorbed by four hours.50 Delayed toxicity can occur after massive ingestions or greater than 25 grams (g), or 300 milligrams/kilogram (mg/kg), due to late or double peaks from delayed gastric emptying.51 When the rate and quantity of N-acetyl-p-benzoquinoneimine (NAPQI, a toxic metabolite) formation overwhelms the supply and turnover of glutathione, the liver is prone to injury because NAPQI can no longer be detoxified (see Figure 2).49-51 The depletion of hepatic glutathione stores occurs in the setting of single or supratherapeutic/chronic overdose. Damaged hepatocytes may further delay toxicity because of the prolonged elimination half-life of APAP.
Figure 2. Acetaminophen Metabolism and N-Acetylcysteine Mechanism of Actions |
Acetaminophen (APAP) metabolism and N-Acetylcysteine (NAC) mechanism of actions (denoted by asterisks). Nontoxic metabolic byproducts of APAP are excreted renally. NAC prevents toxicity by mostly replenishing hepatic glutathione by serving as its precursor, limiting the formation of NAPQI and increasing the capacity to detoxify NAPQI that is formed. NAC also returns part of NAPQI to APAP and enhances sulfation, thereby increasing inactivation and elimination. Parts of molecules were generated using molview.org. |
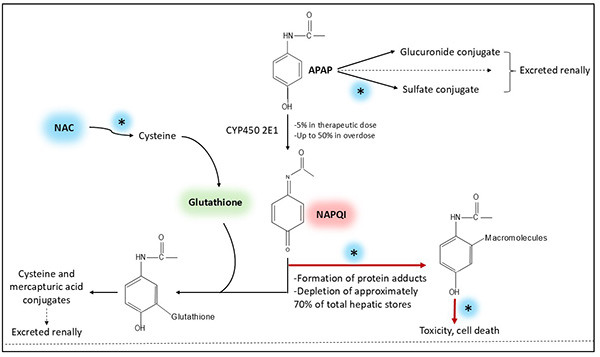 |
Adapted from: Hendrickson RG, McKeown NJ. Acetaminophen. In: Nelson LS, Howland MA, Lewin NA, et al, eds. Goldfrank’s Toxicologic Emergencies. 11th ed. McGraw-Hill Education; 2019:472; and Heard KJ. Acetylcysteine for acetaminophen poisoning. N Engl J Med. 2008;359(3):285-292. |
There are numerous APAP ingestion patterns, and the risk of hepatotoxicity changes in proportion to the amount ingested and duration of exposure.50 The low end of an acute toxic dose of APAP is cited as 7.5 g in adults and 150 mg/kg in children.50 However, approximate doses of at least 150 mg/kg in adults or 200 mg/kg in children typically are needed to cause hepatotoxicity.50 Chronic daily dosing may result in toxicity with thresholds of 6 g/day to 8 g/day in adults and 60 mg/kg/day to 150 mg/kg/day in children.50
During the initial stages of acute APAP toxicity, patients may be asymptomatic or have nonspecific GI symptoms. Elevation of transaminases commonly begins within 24 hours as NAPQI begins to accumulate. Between 72 to 96 hours after ingestion, fulminant hepatic failure may develop with encephalopathy and multiorgan failure. Renal injury may develop between days 2 and 5.49,50 Aspartate aminotransferase (AST) elevation is the most sensitive marker to detect the onset of hepatotoxicity and precedes major hepatic dysfunction. Aminotransferase concentrations above 10,000 IU/L can occur.50 If there is evidence of hepatic failure, monitoring prothrombin time, international normalized ratio, glucose, lactate, and pH are recommended as important determinants of prognosis and treatment.
Knowledge of the time of ingestion, APAP formulation, and concentration are essential for managing APAP toxicity. Patients can be risk stratified using the Rumack-Matthew nomogram for a single acute ingestion if the time of ingestion is known and APAP concentration is measured between four and 24 hours after ingestion. If the time is unknown, use the earliest possible time. For an acute single ingestion, the emergency provider can consider administering activated charcoal within one to two hours of ingestion.50,52 If the concentration is above the treatment line (150 mcg/mL at four hours), the provider should administer N-acetylcysteine (NAC) as soon as possible. Dialysis is not routinely used but is considered in select cases of large ingestions.50,52 Management for APAP ingestions other than a single acute ingestion usually is determined by the patient’s presentation and may differ on a case-by-case basis. Conservative management is recommended for ingestions of sustained-release preparations since the nomogram may be unreliable for risk assessment.53 In these cases, the provider can consider obtaining an additional six- to eight-hour concentration if the initial four-hour APAP level is below the treatment line.52,54 Additionally, the provider also can consider fomepizole as an adjunct for massive APAP ingestions, delayed presentation, and signs of significant hepatotoxicity where NAC may be inadequate.51,55 Treatment of patients with suspected APAP toxicity also should be guided with the assistance of poison control, a toxicologist, and/or a hepatologist if available. Patients with severe suspected toxicity and/or those who are receiving NAC typically require intensive care admission.
Salicylates
With 25.6 million adults 40 years of age or older reporting aspirin use in 2021, it is no surprise that ED physicians encounter many patients who report taking “baby” aspirin.56 Other salicylate-containing products include bismuth subsalicylate and methyl salicylate (i.e., oil of wintergreen; one teaspoon is equivalent to about 21.7 adult aspirin tablets ).57,58 Aspirin’s beneficial properties arise from its irreversible COX 1 and 2 inhibition. The toxic dose of aspirin is cited at 150 mg/kg or 6.5 g.57,58 At toxic levels, salicylates directly stimulate medullary neurons leading to hyperventilation and uncouple oxidative phosphorylation causing a shift toward anaerobic metabolism.
Initial features of acute salicylate toxicity typically occur within one to two hours of ingestion. They include nonspecific GI symptoms, tachypnea, and neurologic symptoms (e.g., tinnitus, drowsiness, headache, and dizziness). However, the onset of symptoms can be significantly delayed up to 24 hours, especially after ingestion of enteric-coated formulation because prolonged retention in the GI tract can slow absorption.57,58 Concomitant renal or hepatic dysfunction and the presence of other coingestants also may contribute to delayed toxicity. As the toxicity progresses, hyperthermia, neuromuscular irritability, and metabolic acidosis develop, with worsening acidosis portending poor prognosis.57,58
Salicylate levels greater than 30 mg/dL commonly are used as a cutoff for diagnosing toxic exposure. Management of salicylate poisoning is focused on supportive care. Table 3 summarizes the goals of management. Providers should consider admission for all symptomatic patients. Therapies are directed at mitigating CNS salicylate exposure, which is a primary site of toxicity. Activated charcoal is strongly recommended within two hours of ingestion in the absence of contraindications.57,58 Well-done studies are lacking, but some clinicians consider whole bowel irrigation or multiple-dose activated charcoal (MDAC) for ER preparations or concerns for pharmacobezoar formation.57,58 If intubation is necessary, sodium bicarbonate administration immediately prior to intubation has been used to prevent a precipitous drop in pH and subsequent accumulation of salicylates in the CNS.57 Drugs that cause respiratory suppression should be avoided when clinically possible. Providers should consider aggressive fluid resuscitation for volume depletion.57 Sodium bicarbonate is recommended for symptomatic patients whose salicylate level is above 40 mg/dL with acidic urine or for clinically suspected cases of toxicity with pending salicylate and blood gas laboratory tests.57 Alkalinizing the urine enhances salicylate excretion. Dextrose is recommended for any altered mental status, but especially if the glucose level is less than 100 mg/dL.57,58 Hemodialysis may be needed in cases of significant toxicity.59
Table 3. Goals of Salicylate Toxicity Management in the Emergency Department Setting |
|
Hypoglycemic Agents
Oral hypoglycemic exposures, particularly sulfonylureas, are more commonly reported to poison control centers compared to insulin exposures.60 Most cases are unintentional because of exploratory ingestions in children or therapeutic errors in adults.61 All forms of insulin (rapid, short, intermediate, or long-acting) and sulfonylureas can cause delayed onset and prolonged hypoglycemia in overdose.62 Large single overdoses of short-acting insulins can mimic overdoses of long-acting insulins likely because of depot effect, in which the large volume of the insulin solution injected significantly reduces absorption by compressing surrounding tissue.61 The extent of delayed onset and duration of hypoglycemia are particularly pronounced after overdoses of long-acting insulin (e.g., glargine, because of its long duration of action of 24 hours) and after sulfonylurea overdose (because of a half-life that approaches 24 hours).60,63 Toxic and lethal doses are dependent on the patient’s baseline glucose level, making the threshold for the onset of severe hypoglycemia different for each patient.64
The primary toxic effects of insulin and sulfonylurea poisonings are hypoglycemia and its secondary insults to the CNS.60,65 Hypoglycemia increases the risk of adverse cardiovascular events, including stress cardiomyopathy and dysrhythmias. Hypoglycemia also can lead to pulmonary edema, hypokalemia, and mild hypothermia.60,64,65 The onset of hypoglycemia can be delayed up to 18 hours after insulin glargine overdose.60,61 However, after a massive insulin glargine overdose, recurrent and prolonged hypoglycemic episodes have been reported to occur up to 192 hours.60,63,66 Prolonged hypoglycemia is thought to occur because of changes in insulin pharmacokinetics, including slowed absorption from the injection site due to mass effect and reduced local blood flow.63,67 In sulfonylurea overdose, the onset of hypoglycemia can range from one to 21 hours, but usually occurs within five to eight hours.60,61 In the pediatric population, even a single tablet ingestion of sulfonylureas can result in delayed onset of hypoglycemia.60 The onset of hypoglycemia in adult overdoses usually is more rapid.61
Point-of-care glucose should be obtained immediately for anyone with a known or suspected hypoglycemic agent toxicity. Hypoglycemia should be considered on the differential for any patient presenting with somnolence, obtundation, or with focal neurologic abnormalities since hypoglycemia may mimic a cerebral vascular accident. Apart from intentional or unintentional toxicity from a hypoglycemic agent, it also is important consider other etiologies and differentials for hypoglycemia, such as sepsis or insulinoma. (See Table 4.) The emergency provider should consider assessing renal and hepatic function since dysfunction of both organs can prolong the half-life of hypoglycemic agents. Furthermore, the addition of serum insulin and C-peptide levels to the diagnostic evaluation can prove helpful if there is a concern for the use of exogenous insulin.61 In insulin poisoning, identifying the insulin analog and dose may be helpful in determining treatment and observation duration. In sulfonylurea overdose, administering activated charcoal can be beneficial in the absence of contraindications.60
Table 4. Differential Diagnosis of Hypoglycemia |
|
The mainstay of insulin and sulfonylurea poisoning is glucose replacement therapy to achieve euglycemia. Usually, the dose of dextrose is selected based on the severity of hypoglycemia and the patient’s clinical status.65 For symptomatic hypoglycemia, administering a dextrose bolus followed by an infusion can be considered.60 One drawback of dextrose administration is the substantial risk for recurrent hypoglycemia, especially in patients with a functional pancreas (e.g., patients without diabetes or with type 2 diabetes) that can produce insulin via glucose-stimulated insulin release.60 In addition, octreotide also may be used to prevent recurrent hypoglycemia.60,61 Octreotide generally is well tolerated without serious adverse effects.60 Glucagon typically is not recommended because it is ineffective in glycogen-depleted states and can stimulate pancreatic insulin release.60 Emergent dialysis typically is not indicated in patients presenting with hypoglycemic agent toxidrome.64,65 The emergency provider should consider admission for patients with intentional overdose of parenteral insulin or sulfonylurea, or unintentional overdose of parenteral long-acting insulin.60 For unintentional sulfonylurea exposure in children, a minimum observation period of approximately 24 hours is recommended.60 One study of oral sulfonylurea ingestions in children found that hypoglycemia typically develops within 13 hours.68
Citalopram and Escitalopram
Compared to older tricyclic antidepressants, selective serotonin reuptake inhibitors (SSRIs) are relatively safer in overdose.23 Some agents, such as citalopram and its isomer escitalopram, warrant further consideration. Citalopram and escitalopram are potent inhibitors of serotonin uptake in the brainstem without inhibiting norepinephrine or dopamine reuptake.23 Because citalopram and escitalopram already have very long elimination half-lives of 33 to 37 hours and 22 to 32 hours, respectively, overdose can contribute to delayed toxicity. Furthermore, the longer half-life of one of their metabolites and its cardiotoxic properties is concerning.69
The most frequently reported clinical effects of excess serotonergic stimulation are drowsiness, nausea, vomiting, tremors, and sinus tachycardia.23,70 Serotonin toxicity may occur in SSRI overdose.71 Interestingly, although the dose increases the probability of serotonin toxicity, it may only have a small effect.71 Among SSRIs, citalopram and escitalopram have significant potential for cardiotoxicity and neurotoxicity. In overdose, citalopram is more likely to cause conduction disturbances than escitalopram and has been associated with up to an eight-fold higher seizure risk.70,72 Citalopram overdoses less than 600 mg often have only mild to moderate neurologic symptoms (e.g., dizziness, somnolence). In contrast, citalopram overdoses exceeding 600 mg are at increased risk for more severe symptoms, including seizures, QT prolongation, QRS prolongation via sodium channel blockage, wide complex ventricular dysrhythmias, and, rarely, death.23,73 The minimum dose of escitalopram needed to cause severe toxic effects is reported to be 300 mg.72 Citalopram and escitalopram both cause QT prolongation in a dose-dependent manner.23 In addition, seizures, electrocardiogram (ECG) changes, and torsades de pointes (TdP) can occur but are uncommon. In citalopram overdose, seizures typically occur early, within two hours of ingestion, while ECG abnormalities can be delayed up to 24 hours after ingestion.23
As part of the diagnostic evaluation of a patient with suspected SSRI overdose, a 12-lead ECG should be performed to assess for evidence of hypokalemia, hypomagnesemia, and hypocalcemia. Management of citalopram and escitalopram overdose is focused on supportive care. The administration of activated charcoal can be considered if the ingestion occurred within four hours of presentation because it may reduce QT prolongation.23 The minimum observation period depends on the ingested amount and whether activated charcoal was administered. Ingestion of more than 600 mg of citalopram or 300 mg of escitalopram requires cardiac monitoring for a minimum of eight hours (11 hours without activated charcoal).74 Ingestion of more than 1,000 mg of citalopram or 500 mg escitalopram requires cardiac monitoring for a minimum 13 hours.74,75 Patients may be medically cleared at the end of the observation period if they are asymptomatic and QT is less than 450 msec.74 Magnesium sulfate and cardiac pacing have been used in the rare cases of SSRI toxicity leading to TdP.76 Sodium bicarbonate by intravenous (IV) push often is considered to treat QRS widening.47 If conduction abnormalities or dysrhythmias persist despite sodium bicarbonate, lidocaine may be used as a second-line antidysrhythmic.77 The provider may treat seizures with traditional therapies, including benzodiazepines, which may be followed by barbiturates if the seizure persists.78,79 It is important to discontinue and avoid any provision of other serotonergic medications if there is suspicion for serotonin toxicity.
Colchicine
The use of colchicine is limited by its narrow therapeutic index and toxicity. Doses above 0.5 mg/kg generally are considered lethal.80,81 Most exposures of colchicine in the United States are unintentional.80 By binding to tubulin, colchicine interferes with cell replication and intracellular transport pathways. It also exerts anti-inflammatory effects, competitively antagonizes gamma-aminobutyric acid type A (GABAA) receptors, and inhibits uric acid crystal deposition.80,82 Colchicine toxicity can present in a delayed fashion as a consequence of cellular mitotic arrest.81 Furthermore, colchicine’s half-life is prolonged up to 32 hours in overdose from four to 16 hours in therapeutic doses (usually 0.5 mg to 3 mg daily).80,82 Colchicine is subject to significant drug interactions, including with diltiazem, verapamil, amiodarone, fluconazole, erythromycin, and grapefruit juice.81
Colchicine toxicity has a prolonged course that progresses through three clinical phases. As well, the effects of colchicine can be dose dependent.80 In the early phase (zero to 24 hours), gastrointestinal (GI) symptoms such as nausea, vomiting, and diarrhea predominate and can lead to significant hypovolemia.80,81 In the mid-phase (one to seven days), multiorgan dysfunction ensues and the bone marrow is significantly affected. Pancytopenia, dysrhythmias, cardiac arrest, sepsis, acute respiratory distress syndrome, rhabdomyolysis, seizures, and metabolic derangements can occur.80,81 The late phase (> 7 days) is the recovery phase for survivors, and symptoms include alopecia, neuropathy, and myopathy.80,81
A colchicine level does not typically correlate with the ingested amount nor symptom severity.80 A concentration above 3 ng/mL generally is associated with toxicity. The mainstay of colchicine poisoning is supportive care. An observation period of at least 24 hours is strongly recommended for anyone with a known or suspected significant colchicine overdose.81 It may be reasonable to perform orogastric lavage within one to two hours of colchicine overdose, followed by activated charcoal, or activated charcoal alone if orogastric lavage is unavailable.80,81 Administration of granulocyte-colony stimulating factor often is considered if leukopenia develops.80 The use of extracorporeal elimination techniques for colchicine toxicity generally is ineffective.83 Hemodialysis can be used if renal failure develops as part of the toxidrome.80,81 In addition, early plasmapheresis may prevent significant toxicity from a lethal overdose.83
Inhaled Toxicants
Inhaled toxicants can be broadly categorized into simple asphyxiants, pulmonary irritants, agents that interfere with red blood cell oxygen transport, chemical asphyxiants (mitochondrial poisons), and other inhaled systemic toxins. The severity of toxicity depends on properties of the agent, exposure circumstances (inhalant concentration, duration of exposure), and health of the exposed individual. Treatment is aimed at limiting further exposure and decontamination, evaluating for co-exposures, and supporting cardiopulmonary status. The two inhaled toxicants discussed in this section are hydrocarbons and phosgene.
Hydrocarbons
Three populations at particular risk for hydrocarbon-related injuries include children with unsupervised ingestions, workers with occupational exposures (plastic, rubber, printers, paints, hazardous wastes), and young adults who intentionally misuse inhalants.84,85 Three common techniques for recreational inhalant misuse include sniffing (direct inhalation from container), huffing (soaking a cloth in the substance and holding it over the nose and mouth), and bagging (breathing vapor directly from a plastic or paper bag).85 Children younger than 6 years of age are disproportionately affected because they can unintentionally aspirate the product.86
Hydrocarbon toxicity can present with delayed pneumonitis with radiographic evidence of pneumonitis developing as early as 15 minutes and up to 24 hours after exposure. Radiographic abnormalities do not often correlate with the severity of pulmonary toxicity.84 Most patients who end up developing pulmonary toxicity have initial coughing, gagging, or choking.84 Halogenated hydrocarbons in particular can cause cardiac dysrhythmias by sensitizing the myocardium to the effects of catecholamines.85 The use of short-acting beta-blockers is recommended for these dysrhythmias.84 Malignant dysrhythmias in the context of inhalant abuse is termed “sudden sniffing death syndrome.”85 Thermal injury to the lips and oropharynx also can occur with halogenated hydrocarbon abuse.87 Hydrocarbons can cause mild transient CNS depression by displacing oxygen from the environment (asphyxiant effect).84
A six-hour observation period followed by a chest X-ray (CXR) is recommended for patients exposed to hydrocarbons.84 The asymptomatic patient with a normal six-hour CXR can be considered for discharge and return precautions.84 The asymptomatic patient with radiographic evidence of pneumonitis can be safely discharged at six hours as long as they can be reevaluated in 24 hours.84 The mildly symptomatic patient whose symptoms quickly resolve after the six-hour period with a normal CXR can be discharged.84 Hospitalization is strongly recommended for patients who have persistent clinical evidence of toxicity, including those with continued symptoms with or without an abnormal CXR.84
Phosgene
Despite being a highly toxic irritant gas and used as a chemical weapon previously, phosgene currently is an indispensable industrial chemical used worldwide to make plastics and pesticides.88,89 Phosgene is known to have a “hay or grass”-like odor and swiftly reacts with alveolar surfactant, binds to amino acids, and releases inflammatory mediators and reactive oxygen.88-90 Given the paucity of human data, the pattern of lethality in humans is unclear.91 However, 30 minutes of exposure to 17 parts per million (ppm) (510 ppm x min) of phosgene has been reported to be lethal.91
Individuals poisoned by phosgene exhibit a range of symptoms within minutes to hours after exposure. Symptoms usually are limited to minor throat, eye, and nose irritation, but decreased oxygen saturation can occur from acute lung injury.90 More moderate to severe symptoms include choking sensation, dyspnea, chest pain, vomiting, and burning sensation in the mouth or throat.90 At the lower to moderate dose and exposure, pulmonary edema occurs within 15-20 hours of exposure.88 The lack of early warning signs combined with an asymptomatic period that can last up to 24 hours may delay the recognition of exposure, contributing to delayed toxicity.91 Treatment of phosgene toxicity is mostly supportive care. Prolonged observation is required for exposed patients, given the potential for delayed pulmonary edema and respiratory failure.89 Supplemental oxygen may be required in patients who develop hypoxia.90 In addition, bronchodilators may be considered to relieve symptoms of bronchoconstriction.90 Empiric antibiotics generally are not recommended unless there is clinical evidence of infection. Moreover, the use of steroids is controversial and is not unanimously recommended.90,92 Lastly, mechanical ventilation, fluid resuscitation, and ECMO may be beneficial in severe cases.90,92
Chlorfenapyr
Of all the xenobiotics discussed here, chlorfenapyr poisoning has the longest reported potential for delayed onset of toxicity, with effects appearing up to 14 days later.93 Although human chlorfenapyr poisonings are geographically concentrated in South and East Asia, it would be prudent for all emergency medicine physicians to understand the clinical course of chlorfenapyr poisoning because of the potential for morbidity and mortality.93 Chlorfenapyr is produced by Streptomyces fumanus bacteria. It is a member of a newer class of pesticides that was registered by the U.S. Environmental Protection Agency in 2001.94 Chlorfenapyr acts as a pro-insecticide that undergoes biotransformation by unspecified hepatic CYP450 enzymes. One of its metabolites is a potent uncoupler of mitochondrial oxidative phosphorylation.93,95 A clear dose-response for chlorfenapyr toxicity has not been established.93 However, death has been reported following oral ingestion of as little as 5 mL of 10% chlorfenapyr.96
The median time to onset of toxic effects was shown to be two hours in survivors and 10 hours in fatalities.93 The long latency period may be due to the time required for toxic metabolites to form. Early toxic effects include nausea, vomiting, diarrhea, diaphoresis, and altered mental status. Tachypnea and tachycardia are the most common vital sign abnormalities on initial presentation.93 Progression of encephalopathy and autonomic instability also are common. Hyperthermia in the later stages is an ominous sign and frequently precedes hemodynamic collapse and death.93,97
Management of chlorfenapyr poisoning is focused on supportive care. Serum levels of chlorfenapyr and its metabolite can be obtained if testing is available. Laboratory markers for clinical severity currently are unknown, but increased creatinine kinase activity is the most reported laboratory abnormality.93 Hyperlactatemia and metabolic acidosis also may be seen. Brain imaging may be considered since abnormal neuroimaging has been consistently reported in patients with chlorfenapyr toxicity.93 Because symptoms may occur days later, it is challenging to determine an appropriate observation time. Nonetheless, the asymptomatic patient should be monitored vigilantly given the considerable potential for rapid deterioration. Treatments can vary considerably and are focused mostly on symptomatology. While data are lacking on human absorption, gastric decontamination can be considered.93 The provider may consider treating hyperthermia with external cooling since hyperthermia induced by uncoupling of oxidative phosphorylation is unlikely to respond to antipyretics.93 Extracorporeal treatment may have little value in chlorfenapyr overdose.93
Mushrooms
Amanita
Cyclopeptide-containing mushroom species, mostly Amanita phalloides, are responsible for 90% to 95% of mushroom-related fatalities.98,99 Amatoxin poisoning’s high mortality rate warrants careful consideration and clinical management. After amatoxin (the most prominent toxin) is rapidly absorbed from the GI tract, it irreversibly inhibits deoxyribonucleic acid (DNA)-dependent ribonucleic acid (RNA) polymerase II, halting protein synthesis.99 Amatoxin poisoning is characterized by a long latency period that typically lasts six to 12 hours but can extend up to 24 hours. It is followed by three phases. In phase I, profuse watery diarrhea and vomiting are seen at five to 24 hours after ingestion. Phase II, the quiescent phase, occurs within 36 hours after ingestion when there may be transient clinical improvement, but GI symptoms persist, and hepatic injury develops. Phase III occurs in severe cases and within two to six days after ingestion, where hepatotoxicity becomes clinically apparent along with AKI, and can culminate in death.99,100
Treatment is focused on supportive care. Symptomatic patients should be admitted due to the possibility of hepatotoxicity leading to death.100 MDAC within 12 to 24 hours after ingestion may be considered.98,101 Liver transplant may be needed in severe cases of liver failure secondary to toxicity.101 There are many potential drug therapies, but none have strong clinical data to support reversal of hepatotoxicity.101 For fulminant hepatic failure, NAC and silibinin (an extract from milk thistle) may be used as they seem to have more pronounced clinical benefit compared to other therapies.98,99,101 Cyclosporin also has been used with the goal of reducing hepatotoxicity.102
Gyromitra
Gyromitra poisonings are rare in the United States but can be significant in a case of mistaken mushroom identification. The primary site of toxicity is the CNS.100 Initially, a nonspecific GI prodrome occurs at five to 10 hours after ingestion. Liver injury can occur over the next few days. In the first 12 to 48 hours, refractory seizures and coma may occur in the most severe poisonings. Treatment is supportive care. Administering activated charcoal in the absence of contraindications may be beneficial as initial therapy.100 Administration of pyridoxine (vitamin B6), in addition to benzodiazepines, may be required to halt seizure activity.78,100,103
Cortinarius
Cortinarius mushrooms can cause severe renal failure with a delay in symptom onset.103 Cortinarius poisoning is characterized by two phases. In phase I, patients experience mild GI irritation anywhere from one day to two weeks after ingestion. In phase II, patients develop AKI due to effects from a toxic metabolite.100,103 Because of the lag phase between ingestion and symptom onset, there is little value in testing blood for toxin. The mainstay of treatment is preventing secondary complications such as kidney failure. Hemodialysis may be necessary in cases of severe toxicity.100 Renal function should be monitored for a prolonged period because of anticipated slow resolution of renal dysfunction.100
Summary
There are numerous ways in which various drugs and toxins can produce delayed toxicity. It is important for the emergency provider to maintain a high degree of clinical suspicion for possible toxidromes because not all delayed toxic effects are related to overt overdoses. There are a multitude of patient and xenobiotic factors that may play a role in cases of inadvertent delayed toxicity. In addition, the development of toxic metabolites may play a significant role in contributing to delayed toxicity. Knowledge of underlying toxic mechanisms, respective management, and therapeutic options along with disposition considerations is critical for the emergency provider in the evaluation of patients with whom there is a concern for delayed toxicity.
Matthew Stripp, MD, is Director of Toxicology, Core Faculty, Emergency Medicine Residency, Allegheny Health Network, Pittsburgh; Assistant Professor, Department of Emergency Medicine, Drexel University College of Medicine, Philadelphia.
Claire Seo, MD, is Emergency Medicine Resident, Allegheny General Hospital, Pittsburgh.
References
1. Howland M. Pharmacokinetic and Toxicokinetic Principles. In: Nelson L, Howland M, Lewin N, et al, eds. Goldfrank’s Toxicologic Emergencies. 11 ed. McGraw-Hill Education; 2019.
2. Holford NH. Pharmacokinetics & Pharmacodynamics: Rational Dosing & the Time Course of Drug Action. In: Katzung B, Vanderah T, eds. Basic and Clinical Pharmacology. 15th ed. McGraw-Hill Education; 2021:42.
3. Janzen KM, Poloyac SM. Clinical Pharmacokinetics and Pharmacodynamics. In: DiPiro JT, Yee GC, Haines ST, et al, eds. DiPiro’s Pharmacotherapy: A Pathophysiologic Approach. 12th ed. McGraw-Hill; 2023.
4. Lehman-McKeeman L. Mechanisms of Toxicity. In: Klaassen C, ed. Casareet & Doull’s Toxicology: The Basic Science of Poisons. 9th ed. McGraw-Hill Education; 2019.
5. Castro DM, Dresser L, Granton J, Fan E. Pharmacokinetic alterations associated with critical illness. Clin Pharmacokinet. 2023;62:209-220.
6. Stillhart C, Asteriadis A, Bocharova E, et al. The impact of advanced age on gastrointestinal characteristics that are relevant to oral drug absorption: An AGePOP review. Eur J Pharm Sci. 2023;187:106452.
7. Lista AD, Sirimaturos M. Pharmacokinetic and pharmacodynamic principles for toxicology. Crit Care Clin. 2021;37:475-486.
8. Zhang W, Zhang Q, Cao Z, et al. Physiologically based pharmacokinetic modeling in neonates: Current status and future perspectives. Pharmaceutics. 2023;15:2765.
9. Ding H. Modified-Release Drug Products and Drug Devices. In: Ducharme MP, Shargel L, eds. Shargel and Yu’s Applied Biopharmaceutics and Pharmacokinetics. 8th ed. McGraw-Hill Education; 2022:305.
10. Drevin G, Malbranque S, Jousset N, et al. Pharmacobezoar-related fatalities: A case report and a review of the literature. Ther Drug Monit. 2023;46(1):1-5.
11. Li YK, Lam KF, Wong CLW, Wong A. In vitro study of pharmacobezoar formation in simulated acetaminophen overdose. Clin Toxicol (Phila). 2020;58(9):900-906.
12. Inoue F, Okazaki Y, Huh K, et al. Pharmacobezoar associated prolonged clinical course in a patient with immediate release quetiapine overdose. J Med Toxicol. 2024;20:430-433.
13. Hoegberg LCG. Techniques Used to Prevent Gastrointestinal Absorption. In: Nelson LS, Howland MA, Lewin NA, et al, eds. Goldfrank’s Toxicologic Emergencies. 11th ed. McGraw-Hill Education; 2019:48.
14. Johnson AR, Brown KM. Principles of Clinical Toxicology. In: Brunton L, Knollmann B, eds. Goodman & Gillman’s: The Pharmcological Basis of Therapeutics. 14th ed. McGraw-Hill Education; 2023:155.
15. Yamamoto T, Malavsai E, Archer JR, et al. Management of body stuffers presenting to the emergency department. Eur J Emerg Med. 2016;23(6):425-429.
16. Traub SJ, Hoffman RS, Nelson LS. Body packing — the internal concealment of illicit drugs. N Engl J Med. 2003;349:2519-2526.
17. Puntonet J, Gorgiard C, Soussy N, et al. Body packing, body stuffing and body pushing: Characteristics and pitfalls on low-dose CT. Clin Imaging. 2021;79:244-250.
18. Dufayet L, Deguette C, Vodovar D. Emergency medicine management of patients transporting cocaine by body-packing. Eur J Emerg Med. 2023;30:161-162.
19. deBakker JK, Nanayakkara PWB, Geeraedts LMG, et al. Body packers: A plea for conservative treatment. Langenbecks Arch Surg. 2012;397:125-130.
20. Rose SR, Cumpston KL, Kim J, et al. Absorption of salicylate powders versus tablets following overdose: A poison center observational study. Clin Toxicol (Phila). 2016;54(9):857-861.
21. Gummin DD, Mowry JB, Beuhler MC, et al. 2021 Annual Report of the National Poison Data System© (NPDS) from America’s poison centers: 39th annual report. Clin Toxicol (Phila). 2022;60(12):1381-1643.
22. Pope JD, Drummer OH, Schneider HG. False-positive amphetamines in urine drug screens: A 6-year review. J Anal Toxicol. 2023;47:263-270.
23. Stork CM. Serotonin Reuptake Inhibitors and Atypical Antidepressants. In: Nelson LS, Howland MA, Lewin NA, et al, eds. Goldfrank’s Toxicologic Emergencies. 11th ed. McGraw-Hill Education; 2019:1054.
24. El Mansari M, Manta S, Oosterhof C, et al. Restoration of serotonin neuronal firing following long-term administration of bupropion but not paroxetine in olfactory bulbectomized rats. Int J Neuropsychopharmacol. 2015;18(4):1-8.
25. Offerman S, Gosen J, Thomas SH, et al. Bupropion associated seizures following acute overdose: Who develops late seizures. Clin Toxicol (Phila). 2020;58(12):1306-1312.
26. Stewart E, Grewal K, Hudson H, et al. Clinical characteristics and outcomes associated with bupropion overdose: A Canadian perspective. Clin Toxicol (Phila). 2020;58(8):837-842.
27. Idowu D, Ezema K, Corcoran J, Farkas A. The predictive value of heart rate in determining clinical course after a bupropion overdose. Clin Toxicol (Phila). 2024;62(5):296-302.
28. Rianprakaisang TN, Prather CT, Lin AL, et al. Factors associated with seizure development after bupropion overdose: A review of the toxicology investigators consortium. Clin Toxicol (Phila). 2021;59(12):1234-1238.
29. Caillier B, Pilote S, Castonguay A, et al. QRS widening and QT prolongation under bupropion: A unique cardiac electrophysiological profile. Fundam Clin Pharmacol. 2011;26(5):599-608.
30. Beuhler MC, Spiller HA, Sasser HC. The outcome of unintentional pediatric bupropion ingestions: A NPDS database review. J Med Toxicol. 2010;6:4-8.
31. Bornstein K, Montrief T, Parris MA. Successful management of adolescent bupropion overdose with intravenous lipid emulsion therapy. J Pediatr Intensive Care. 2019;8:242-246.
32. Bupropion hydrochloride. In: Merative Micromedex® DRUGDEX® (electronic version).
33. Wu P, Jurrlink D. Bupropion. In: Brent J, Burkhart K, Dargan P, et al, eds. Critical Care Toxicology: Diagnosis and Management of the Critically Poisoned Patient. 2nd ed. Springer International Publishing; 2016.
34. Simpson M, Johnson L, Goldfine C. Sodium bicarbonate treatment for QRS widening in bupropion overdoses. Clin Toxicol (Phila). 2023;61(6):436-444.
35. Robinson S. Treatment of status epilepticus and prolonged QT after massive intentional bupropion overdose with lidocaine. Am J Emerg Med. 2022;55:232.e3-232.e4.
36. Heise CW, Skolnik AB, Raschke RA, et al. Two cases of refractory cardiogenic shock secondary to bupropion successfully treated with veno-arterial extracorporeal membrane oxygenation. J Med Toxicol. 2016;12:301-304.
37. O’Brien ME, Chary M, Moonsamy P, et al. Successful use of ECMO and lipid emulsion for massive bupropion overdose: A case report. Toxicology Communications. 2021;5(1):85-87.
38. Gallagher N, Edwards FJ. The diagnosis and management of toxic alcohol poisoning in the emergency department: A review article. Adv J Emerg Med. 2019;3(3):e28.
39. Hoyte C, Schimmel J, Hadianfar A, et al. Toxic alcohol poisoning characteristics and treatments from 2000 to 2017 at a United States regional poison center. Daru. 2021;29:367-376.
40. Ghannoum M, Roberts D, Bouchard J. Enhanced Elimination of Poisons. In: Yu ASL, Chertow GM, Luyckx V, et al, eds. Brenner and Rector’s The Kidney. 11th ed. Elsevier; 2020.
41. National Library of Medicine. PubChem Compound Summary for CID 887, Methanol. https://pubchem.ncbi.nlm.nih.gov/compound/Methanol
42. Kraut J, Mullins M. Toxic alcohols. N Engl J Med. 2018:270-280.
43. Wiener SW. Toxic Alcohols. In: Nelson LS, Howland MA, Lewin NA, et al, eds. Goldrank’s Toxicologic Emergencies. 11th ed. McGraw-Hill Education; 2019:1421.
44. Stašinskis R, Stašinska K, Mukāns M, et al. Changes in ionized calcium in ethylene glycol poisoning. Proc (Bayl Univ Med Cent). 2022;35(4):460-465.
45. Hodgman MJ, Marraffa JM, Wiener BG, et al. Assessing the role of initial serum calcium concentration in patients with ethylene glycol poisoning. J Med Toxicol. 2023;19:368-373.
46. Charney AN, Hoffman RS. Fluid, Electrolytes, and Acid-Base Principles. In: Nelson LA, Howland MA, Lewin NA, et al, eds. Goldfrank’s Toxicologic Emergencies. 11th ed. McGraw-Hill Education; 2019:189.
47. Wax PM, Haynes A. Sodium Bicarbonate. In: Nelson LS, Howland MA, Lewin NA, et al, eds. Goldfrank’s Toxicologic Emergencies. 11th ed. McGraw-Hill Education; 2019:567.
48. Hughes A, Amaducci A, Campleman SL, et al. The toxicology investigators consortium 2023 annual report. J Med Toxicol. 2024;20(4):350-380.
49. McGill MR, Hinson JA. The development and hepatotoxicity of acetaminophen: Reviewing over a century of progress. Drug Metab Rev. 2020;52(4):472-500.
50. Hendrickson RG, McKeown NJ. Acetaminophen. In: Nelson LS, Howland MA, Lewin NA, et al, eds. Goldfrank’s Toxicologic Emergencies. 11th ed. McGraw-Hill Education; 2019:472.
51. Janković SM. Acetaminophen toxicity and overdose: Current understanding and future directions for NAC dosing regimens. Expert Opin Drug Metab Toxicol. 2022;18(11):745-753.
52. Dart RC, Mullins ME, Matoushek T, et al. Management of acetaminophen poisoning in the US and Canada: A consensus statement. JAMA Netw Open. 2023;6(8):e2327739.
53. Spyker DA, Dart RC, Yip L, et al. Population pharmacokinetic analysis of acetaminophen overdose with immediate release, extended release and modified release formulations. Clin Toxicol (Phila). 2022;60(10):1113-1121.
54. Chiew AL, Buckley NA. Acetaminophen poisoning. Crit Care Clin. 2021;37(3):543-561.
55. Kang AM, Padilla-Jones A, Fisher ES, et al. The effect of 4-methylpyrazole on oxidative metabolism of acetaminophen in human volunteers. J Med Toxicol. 2020;16:169-176.
56. Gupta M, Gulati S, Nasir K, Sarraju A. Aspirin use prevalence for cardiovascular disease prevention among U.S. adults from 2012 to 2021. Ann Intern Med. 2024;177(8):1139-1141.
57. Palmer BF, Clegg DJ. Salicylate toxicity. N Engl J Med. 2020;382:2544-2555.
58. Lugassy DM. Salicylates. In: Nelson LS, Howland MA, Lewin NA, et al, eds. Goldfrank’s Toxicologic Emergencies. 11th ed. McGraw-Hill Education; 2019:555.
59. The Extracorporeal Treatments in Poisoning Workgroup. Salicylates. Published 2015. https://www.extrip-workgroup.org/salicylates
60. Bosse GM. Antidiabetics and Hypoglycemics/Antiglycemics. In: Nelson LS, Howland MS, Lewin NA, et al, eds. Goldfrank’s Toxicologic Emergencies. 11th ed. McGraw-Hill Education; 2019:694.
61. Klein-Schwartz W, Stassinos GL, Isbister GK. Treatment of sulfonylurea and insulin overdose. Br J Clin Pharmacol. 2015;81(3):496-504.
62. Moyns EJ, Ferner RE. Treatment of insulin poisoning: A 100-year review. Diabet Med. 2023;40:e15076.
63. Maillot J, Poyat C, Salvadori A, et al. Long acting insulin glargine overdose, a surprising long lifetime. Toxicology Communications. 2019;3(1):30-32.
64. Rzepczyk S, Dolińska-Kaczmarek K, Uruska A, Żaba C. The other face of insulin-overdose and its effects. Toxics. 2022;10(3):123.
65. Baumgartner K, Devgun J. Toxicology of medications for diabetes mellitus. Crit Care Clin. 2021;37:577-589.
66. Lu M, Inboriboon PC. Lantus insulin overdose: A case report. J Emerg Med. 2011;41(4):374-377.
67. Arbouche N, Walch A, Raul JS, Kintz P. Intentional overdose of glargine insulin: Determination of the parent compound in postmortem blood by LC-HRMS. J Forensic Sci. 2023;68:1077-1083.
68. Levine M, Ruha AM, Lovecchio F, et al. Hypoglycemia after accidental pediatric sulfonylurea ingestions. Pediatr Emerg Care. 2011;27(9):846-849.
69. Schreffler SM, Marraffa JM, Stork CM, Mackey J. Sodium channel blockade with QRS widening after an escitalopram overdose. Pediatr Emerg Care. 2013;29(9):998-1001.
70. Yilmaz Z, Ceschi A, Rauber-Lüthy C, et al. Escitalopram causes fewer seizures in human overdose than citalopram. Clin Toxicol (Phila). 2010;48(3):207-212.
71. Cooper J, Duffull SB, Isbister GK. Predicting serotonin toxicity in serotonin reuptake inhibitor overdose. Clin Toxicol (Phila). 2023;61(1):22-28.
72. Hayes BD, Klein-Schwartz W, Clark RF, et al. Comparison of toxicity of acute overdoses with citalopram and escitalopram. J Emerg Med. 2010;39(1):44-48.
73. Kraai EP, Seifert SA. Citalopram overdose: A fatal case. J Med Toxicol. 2015;11:232-236.
74. Citalopram. Merative Micromedex® DRUGDEX® (electronic version).
75. van Gorp F, Duffull S, Hackett LP, Ibsister GK. Population pharmacokinetics and pharmacodynamics of escitalopram in overdose and the effect of activated charcoal. Br J Clin Pharmacol. 2012;73(3):402-410.
76. Thomas SH, Behr E. Pharmacological treatment of acquired QT prolongation and torsades de pointes. Br J Clin Pharmacol. 2015;81(3):420-427.
77. Juurlink DN. Antipsychotics. In: Nelson LS, Howland MA, Lewin NA, et al, eds. Goldfrank’s Toxicologic Emergencies. 11th ed. McGraw-Hill Education; 2019:1032.
78. Kienitz R, Kay L, Beuchat I, et al. Benzodiazepines in the management of seizures and status epilepticus: A review of routes of delivery, pharmacokinetics, efficacy, and tolerability. CNS Drugs. 2022;36(9):951-975.
79. Jain P, Aneja S, Cunningham J, et al. Treatment of benzodiazepine-resistant status epilepticus: Systematic review and network meta-analyses. Seizure. 2022;102:74-82.
80. Santos CD, Schier JG. Colchicine, Podophyllin, and the Vinca Alkaloids. In: Nelson LS, Howland MA, Lewin NA, et al, eds. Goldfrank’s Toxicologic Emergencies. 11th ed. McGraw-Hill Education; 2019:501.
81. Stamp LK, Horsley C, Karu LT, et al. Colchicine: The good, the bad, the ugly and how to minimize the risks. Rheumatology (Oxford). 2024;63:936-944.
82. DynaMed. General Drug Monograph: Colchicine. https://www.dynamed.com/drug-monograph/colchicine
83. Toksul İH, Altundağ İ, Çete R, et al. Successful management of a colchicine overdose with plasmapheresis: A case report. Toxicology Communications. 2023;7(1):2286391.
84. Riggan MAA, Gummin DD. Hydrocarbons. In: Nelson LS, Howland MA, Lewin NS, et al, eds. Goldfrank’s Toxicologic Emergencies. 11th ed. McGraw-Hill Education; 2019:1409.
85. Tormoehlen LM, Tekulve KJ, Nañagas KA. Hydrocarbon toxicity: A review. Clin Toxicol (Phila). 2014;52(5):479-483.
86. Jolliff HA, Fletcher E, Roberts KJ, et al. Pediatric hydrocarbon-related injuries in the United States: 2000-2009. Pediatrics. 2013;131(6):1139-1147.
87. Katz J, Dorey A. Thermal injury after “huffing” compressed air duster: A case report. Toxicology Communications. 2024;8(1):2380932.
88. Pauluhn J. Phosgene inhalation toxicity: Update on mechanisms and mechanism-based treatment strategies. Toxicology. 2021;450:152682.
89. Suchard JR. Chemical Weapons. In: Nelson LS, Howland MA, Lewin NA, et al, eds. Goldfrank’s Toxicologic Emergencies. 11th ed. McGraw-Hill Education; 2019:1741.
90. Asgari A, Parak M, Nourian YH, Ghanei M. Phosgene toxicity clinical manifestations and treatment: A systematic review. Cell J. 2024;26(2):91-97.
91. Hobson ST, Richieri RA, Parseghian MH. Phosgene: Toxicology, animal models, and medical countermeasures. Toxicol Mech Methods. 2021;31(4):293-307.
92. Lu Q, Huang S, Meng X, et al. Mechanism of phosgene-induced acute lung injury and treatment strategy. Int J Mol Sci. 2021;22:10933.
93. Comstock GT, Nguyen HV, Bronstein A, Yip L. Chlorfenapyr poisoning: A systematic review. Clin Toxicol (Phila). 2024;62(7):412-424.
94. United States Environmental Protection Agency. Pesticide Fact Sheet: Chlorfenapyr. https://www3.epa.gov/pesticides/chem_search/reg_actions/registration/fs_PC-129093_01-Jan-01.pdf
95. Holland MG. Insecticides: Organic Chlorines, Pyrethrins/Pyrethroids, and Insect Repellents. In: Nelson LS, Howland MA, Lewin NA, et al, eds. Goldfrank’s Toxicologic Emergencies. 11th ed. McGraw-Hill Education; 2019:1514.
96. Gong Y, Meng QB, Liu L, et al. [Article in Chinese]. [Vigilance against a highly lethal insecticide chlorfenapyr poisoning (report of 4 cases and literature review)]. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. 2021;39(9):689-693.
97. Chung MJ, Mao YC, Hsu CT, et al. A fatal case of chlorfenapyr poisoning and the therapeutic implications of serum chlorfenapyr and tralopyril levels. Medicina (Kaunas). 2022;58:1630.
98. Trakulsrichai S, Sriapha C, Tongpoo A, et al. Clinical characteristics and outcome of toxicity from Amanita mushroom poisoning. Int J Gen Med. 2017;10:395-400.
99. Lecot J, Cellier M, Courtois A, et al. Cyclopeptide mushroom poisoning: A retrospective series of 204 patients. Basic Clin Pharmacol Toxicol. 2023;132:528-537.
100. Goldfrank LR. Mushrooms. In: Nelson LS, Howland MA, Lewin NA, et al, eds. Goldfrank’s Toxicologic Emergencies. 11th ed. McGraw-Hill Education; 2019:1581.
101. Diaz JH. Amatoxin-containing mushroom poisonings: Species, toxidromes, treatments, and outcomes. Wilderness Environ Med. 2018;29:111-118.
102. Mackenzie CA, Austin E, Thompson M, Tirona RG. Cyclosporine as a novel treatment for amatoxin-containing mushroom poisoning: A case series. Toxicology Communications. 2022;6(1):23-27.
103. Dinis-Oliveira RJ, Soares M, Rocha-Pereira C, Carvalho F. Human and experimental toxicology of orellanine. Hum Exp Toxicol. 2016;35(9):1016-1029.
Drug overdose is a common chief complaint in the emergency department. Overdose of certain medications or ingestions of toxins should prompt the emergency physician to carefully deliberate on the appropriate next steps and disposition. Furthermore, delayed toxicity may be under-recognized, and delayed management can lead to life-threatening complications, such as arrhythmias and seizures. Managing the poisoned patient can be challenging because clinical effects often are difficult to predict in circumstances that produce toxicity.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.