An Updated Review on Metabolic Regulation in the Alzheimer’s Brain: Type 3 Diabetes?
EXECUTIVE SUMMARY
The prevalence of Alzheimer’s disease (AD) and type 2 diabetes mellitus (T2DM) both are increasing at an alarming rate.
- Multiple studies have shown the relationship of the two diseases through similar pathological disturbances, such as mitochondrial damage, insulin resistance, inflammation, and cell death.
- The worldwide economic burden of AD and T2DM surpasses trillions of dollars and is expected to double by the end of the next decade.
- Diabetes is a strong risk factor for the development of AD, since more than 70% of patients with AD have either diabetes or some sort of glucose metabolism impairment.
- A main driver of insulin resistance is believed to start with defects in the mitochondria, destabilizing the metabolism of glucose to adenosine triphosphate, with increased production of reactive oxidative species with development of advanced glycation end products.
- Hypometabolism is caused by insulin resistance and mitochondrial dysfunction, mostly localized to the temporal and parietal lobes in the AD brain, leading to impaired glucose transport through decreased glucose transporter type 1 channels, beta amyloid plaques that destabilize synapses and affect the mitochondria, mitochondrial dysfunction that causes inflammation and oxidative stress, and alteration in cerebrovascular blood flow.
- Treatment should be targeted to promoting mitochondrial health through moderate-intensity aerobic exercise; multiple drugs, such as metformin, Szeto-Schiller 31 peptide, coenzyme Q, and imeglimin; a ketogenic diet; and the Mediterranean-DASH Intervention for Neurodegenerative Delay diet.
Introduction
The prevalence of Alzheimer’s disease (AD) is increasing at an alarming rate. While multiple factors, such as increasing life expectancy and a growing aging population, have contributed to an increase in the diagnosis of AD, an important factor — metabolic health — has been identified as a potential determinant. Recently, multiple studies have revealed the relationship between AD and type 2 diabetes mellitus (T2DM) through similar pathological disturbances, such as mitochondrial damage, insulin resistance, inflammation, and cell death. This review will reveal how AD is a disorder of metabolic regulation and will place an emphasis on treatment modalities that target one of the culprits of this disease: mitochondria.
Case Scenario
A 59-year-old female with a past medical history of T2DM presents to your office with concerns about having a prevalent family history of Alzheimer’s dementia. She states that her mother is being taken care of at a nursing facility, which has taken a significant mental, emotional, physical, and financial toll on her and her family. Upon further questioning, you discover that the patient’s mother also has T2DM, which she developed in her 40s. She started developing cognitive decline in her 70s and ultimately was diagnosed with AD almost a decade later.
As per your patient, you continually have recommended that she exercise and eat a healthy diet rich in fruits and vegetables, with a regimen of metformin, 500 mg twice per day. Although your patient has tried to implement these changes in her life, she admits, “I can eat healthy for a few days but then I fall into my old patterns.” She enjoys eating out with her husband as well as going on walks. She walks for about 20 minutes per day. She does not engage in any other type of exercise, including but not limited to strength training, resistance training, or aerobic exercise. Her most recent hemoglobin A1c (HbA1c) is 7. She wishes to “take control” of her health by instituting more lifestyle approaches with the goal of reversing her T2DM as well as preventing the chance of developing memory issues in the future.
You decide to educate her on the importance of controlling her metabolic health to decrease the probability of developing cognitive decline in the future. In addition, as her provider, you ask yourself what types of treatment you could implement into her current treatment regimen that would positively affect her metabolic and brain health.
An Overview of Diabetes and Alzheimer’s Disease
Recent studies reveal an exponential increase in metabolic disorders related to diabetes and obesity.1,2 Increases in caloric intake, excess carbohydrates, and a sedentary lifestyle have greatly contributed to metabolic derangements, namely insulin resistance as seen in T2DM and AD.1,3-6
It is projected that an additional 200 million people will be affected by T2DM by the year 2040, increasing its prevalence to 600 million individuals worldwide.1 (See Figure 1.) Interestingly, the prevalence of AD and other cognitive diseases also has been on the rise, with a projected 139 million individuals affected by the year 2050.2
Figure 1. Projected Prevalence of T2DM vs. Alzheimer’s Disease from 2020 to 2040 |
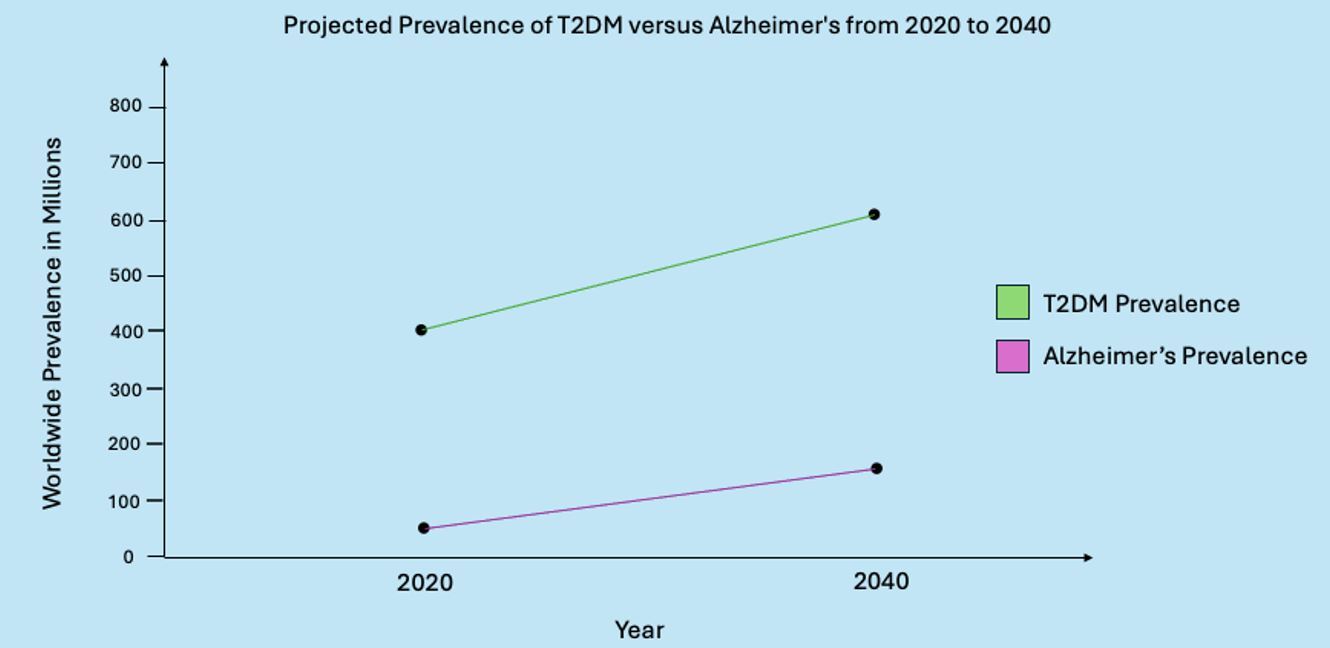 |
T2DM: type 2 diabetes mellitus Created by Gabriela De Los Santos, MD. Adapted from Goyal R, Singhal M, Jialal I. Type 2 Diabetes. StatPearls Publishing; 2024 Jan-. https://www.ncbi.nlm.nih.gov/books/NBK513253/ and Li X, Feng X, Sun X, et al. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990-2019. Front Aging Neurosci 2022;14:937486. |
The worldwide economic burden for T2DM and AD surpasses trillions of dollars and is expected to double by the end of the next decade.2 However, it is important to note that the increase in the diagnosis of AD can partly be attributed to the increase in general life expectancy and an increase in the aging population.7
Decades of research describe the pathophysiology of AD involving the accumulation of amyloid plaques in extracellular compartments leading to damage of synapses.4,8 Accumulation of amyloid plaques is theorized to drive the hyperphosphorylation of intracytoplasmic tau proteins, forming fibrillary tangles and destabilizing microtubular intracellular neuronal transport.8 Although studies have elucidated that the accumulation of amyloid plaques can be genetically driven by an increase apolipoprotein E (APOE) e4 expression, they fail to stress how environmental and lifestyle factors greatly contribute to both the pathophysiology of amyloid development and cognitive decline in AD.
Diabetes is a strong risk factor for the development of AD given the mitochondrial, vascular, and inflammatory modalities that contribute to its progression.9 Vast evidence has elucidated that more than 70% of patients with AD have either diabetes or some sort of glucose metabolism impairment.4,9
Other longitudinal studies have suggested that, compared to patients without diabetes, patients with diabetes have a 65% higher risk of developing AD or other pathological cognitive decline, highlighting a strong relationship between these two metabolic ailments.4,7,9 Therefore, it is vital to understand the pathophysiology of these two diseases as they relate to insulin resistance and mitochondrial dysfunction. Exposing a metabolic relationship between these diseases can provide further understanding of metabolic health and how treatment of insulin resistance can decrease disease burden. These mitochondrial disturbances that are caused by insulin resistance can help researchers develop treatments centralized on preventive medicine.
Part 1: Pathophysiology of End Organ Damage in Diabetes
T2DM is characterized by abnormal carbohydrate metabolism with resultant hyperglycemia. This is associated with hyperinsulinemia, potentiating a dysfunction in peripheral cell insulin receptors.10 Many individuals with T2DM have increased abdominal adiposity and body mass index compared to those without diabetes.1 This presence of abdominal obesity increases circulating inflammatory factors, such as adipokines. In turn, peripheral adipose tissue increases the release of free fatty acids (FFAs), which inhibit muscular and hepatic insulin-mediated glucose uptake and glycogen synthesis.10 This mechanism is known to worsen insulin resistance. In addition, FFAs have been shown to activate inflammatory pathways leading to low-grade inflammation throughout the body.10 This type of inflammation also is associated with negatively influencing the permeability of the gut microbiome, which is intricately connected to the nervous system.1
A main driver of the development of insulin resistance in patients with T2DM is thought to start with defects in the mitochondria throughout tissues.10-13 Persistently elevated blood glucose levels destabilize glucose metabolism to adenosine triphosphate (ATP) in the mitochondria, leading to organelle dysfunction.6,11,12 As a result, there is a decreased production of ATP and an increased production in reactive oxidative species (ROS) seen in beta cells of the mitochondria of the pancreas. Similar mitochondrial changes have been seen in tissues of the liver, muscle, brain, and adipose tissue.11
As tissues continue to become insulin-resistant, the production of proinflammatory cytokines worsens metabolic signaling. An increase in ROS from mitochondrial dysfunction leads to the activation of serine kinases that phosphorylate proteins associated with insulin resistance.13
ROS also increases proinflammatory cytokines that decrease tissue insulin sensitivity. A continual alteration in mitochondrial function negatively alters the mitochondria’s capacity to process fatty acids. Accumulation of fatty acids and diacylglycerol are theorized to activate protein kinase C, leading to lipid-induced mitochondrial dysfunction.13 These fatty acids also travel outside the cell, increasing the production of proinflammatory cytokines that bind to proteins that contribute to impaired insulin signaling. In essence, a state of low-grade inflammation is a clear contributor to the development of insulin resistance with concurrent mitochondrial dysfunction.
The development of advanced glycation end products (AGE) is driven by persistent hyperglycemia as the result of tissue insulin resistance.4,7,9,11-14 (See Figure 2.) This occurs when excess glucose forms covalent bonds with plasma proteins. These new bonds alter enzymatic activity, increase the release of free radicals, and increase the release of proinflammatory cytokines, exacerbating the already present low-grade inflammation.14
Figure 2. The Development of AGEs and its Consequences |
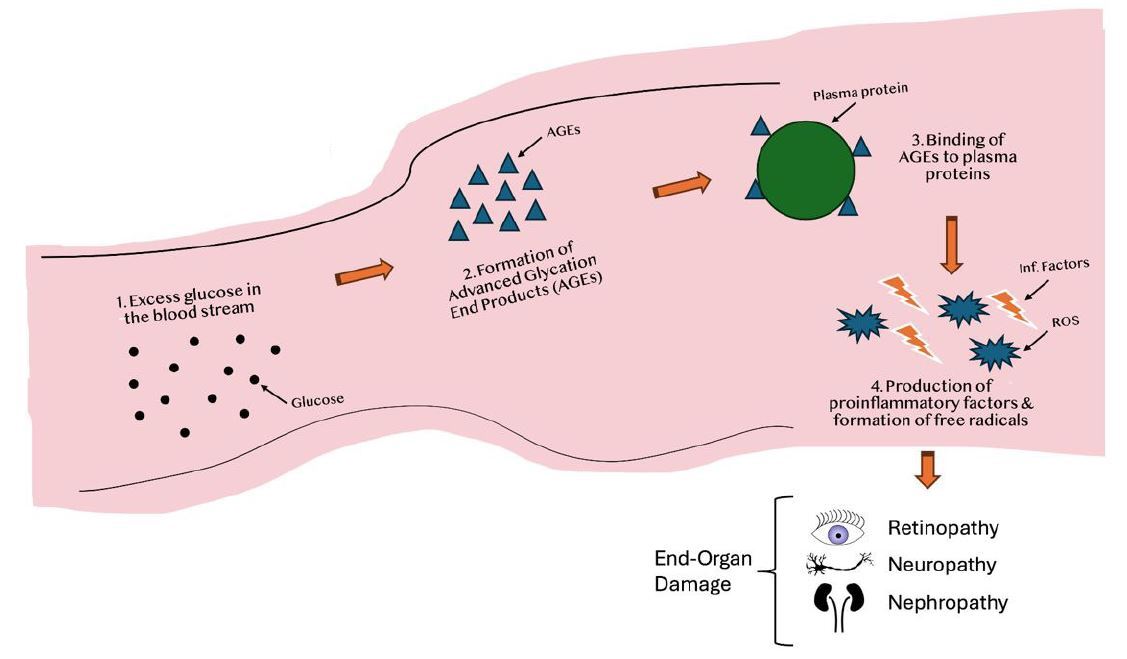 |
AGEs: advanced glycation end products; ROS: reactive oxidative species Created by Gabriela De Los Santos, MD. Not drawn to scale. |
The production of AGEs significantly contributes to the end organ damage seen in diabetes, such as retinopathy, nephropathy, and neuropathy. Unfortunately, these AGEs are resistant to the action of proteinases that theoretically would serve to destabilize their presence.14 Vascular endothelial cells also are susceptible to the damage from AGEs by affecting the brain-blood barrier and contributing to endothelial abnormalities in the brain, thus increasing the incidence of strokes and brain-hypoxic episodes in patients with diabetes.14
In addition, AGEs increase oxidative stress and the production of free radicals that continue to damage subsequent cells in the body, such as neurons.14 AGEs also are known to disturb neuronal connections by accumulating in the structural components of neurofilaments and tubulin, which are imperative for axonal transport.11,12,14 This leads to loss of the fiber as well as subsequent demyelination peripherally, leading to diabetic autonomic and peripheral neuropathy. AGEs also increase reactive oxygen species that glycosylate the enzyme Na+/K+-ATPase, which is involved in the conduction velocity of the nerve, decreasing motor velocity during axonal transport.3,5,6,11,14
Damage to both the endothelial and endoneurial blood vessels is worsened by insulin resistance and hyperglycemia. AGEs are known to bind to the components of the basement membrane, leading to increased thickening and permeability of the vessel.14 This is further exacerbated by a decreased production of nitric oxide (NO) through the accumulation of AGEs. As a potent vasodilator, NO increases blood flow inside vessels. Decreasing NO inherently causes blood vessels to become stiff, which in turn reduces blood flow and increases hypoxic episodes that affect brain parenchyma.11,12,14 A combination of insulin resistance and mitochondrial dysfunction increases the production of ROS, worsening the cellular and structural capabilities of the vascular endothelium.11 With these known facts, neuronal damage can be prevented through tight glucose control, since it reduces the production of AGEs.14
One manifestation of T2DM is the deposition of amyloid plaque in pancreatic beta cells. Similarly, extracellular deposits of amyloid also occur in the pathogenesis of AD.3,4 Extracellular amyloid in the pancreas progresses because of an overproduction of islet amyloid polypeptide (IAPP), a precursor molecule that is co-secreted with insulin.10,14,15 As insulin production increases to achieve glucose homeostasis in the early stages of diabetes, IAPP production also is increased for carbohydrate processing. The development of pancreatic islet amyloid deposits is believed to develop after beta cell exhaustion, leading to a decrease in insulin production by these cells.15 As the result of overproduction and failure of proper processing of IAPP, toxic fibrils of amylin induce apoptosis in the pancreatic beta cells through the formation of pores in the cell membrane. This causes an imbalance of ions across the cell membrane.15 Furthermore, a prolonged state of hyperglycemia also incites the production of IAPP. A similar pathogenesis of amyloid deposit in the form of IAPP in AD has been manifested, which will be discussed in a subsequent section of this review.
Part 2: Pathophysiology of Neurological Damage in Alzheimer’s Disease
The brain’s main source of energy is glucose. Glucose enters the brain by facilitated diffusion via various glucose transporters to be used by neural cells for ATP production. It has been well established that there is a decrease in glucose use, a hypometabolism, caused by insulin resistance and mitochondrial dysfunction mostly localized to the temporal and parietal lobes in the AD brain.3 Moreover, the metabolic pathogenesis of AD has elucidated key products of insulin resistance: impaired glucose transport through decreased glucose transporter type 1 (GLUT1) channels, beta amyloid plaques that destabilize synapses and affect the mitochondria, mitochondrial dysfunction that causes inflammation and oxidative stress, and alteration in cerebrovascular blood flow.3,13,14
Insulin resistance causes decreased glucose uptake and hypometabolism in key brain areas. (See Figure 3.) During a state of hyperglycemia, there first is an upregulation of membrane glucose transporters. However, this soon leads to a negative feedback loop where the production of glucose channels decreases, leading to cellular insulin resistance.16 Hypometabolism in the Alzheimer’s brain is seen mainly in the temporal and parietal lobes, mostly affecting glucose-hungry tissues located in the entorhinal cortex — mainly the hippocampal area.3,17 A decrease in GLUT1 and GLUT3 channels has been observed in Alzheimer’s brains, decreasing glucose transport and leading to neuronal damage from lack of energy sources.17
Figure 3. The Development and Effects of Insulin Resistance in the Brain |
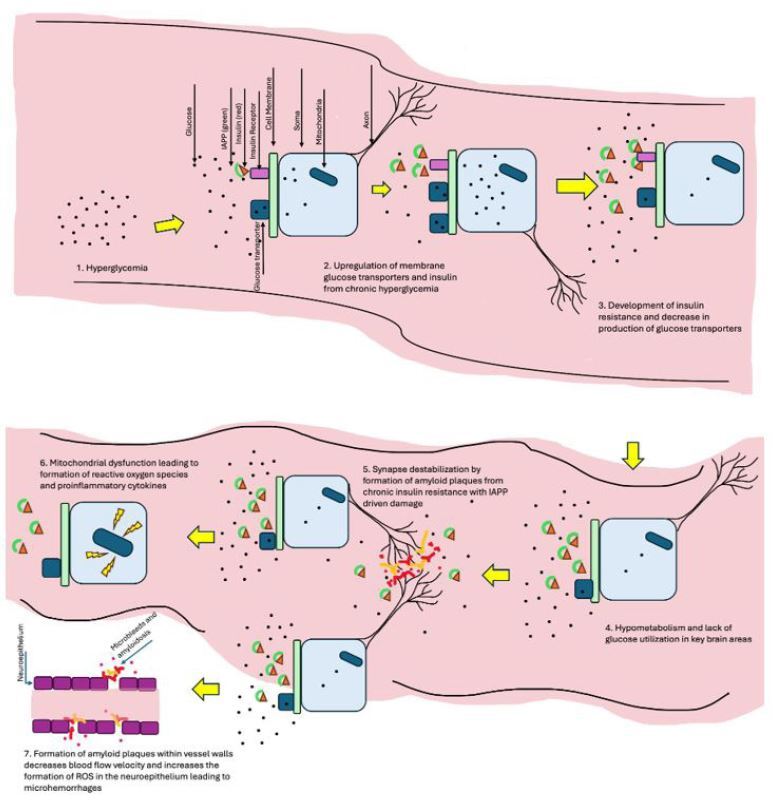 |
IAPP: islet amyloid polypeptide; ROS: reactive oxidative species Created by Gabriela De Los Santos, MD. Not drawn to scale. |
It is hypothesized that decreased glucose processing in these brain areas is related to accumulating amyloid plaques. The brain has a high volume of binding sites for IAPP.15 Therefore, because of insulin resistance, there is a concurrent increase in the release of insulin and IAPP, as discussed previously, which leads to the formation of amylin and amyloid deposits in the brain.4 This key similarity further elucidates the relationship between T2DM and AD — insulin resistance and mitochondrial dysfunction playing a key role in the pathogenesis of both. Amylin deposits have been found in brain endothelium and vasculature, which leads to worsened cognition and cerebral microbleeds, observed in both AD and vascular dementia.4,5
Defects in the vascularity of the brain and increased blood-brain barrier permeability are seen in AD and other dementias. Risk factors such as hyperinsulinemia, diabetes, and hypertension worsen brain vascularity.5 The formation of amyloid plaques within vessel walls decreases blood flow velocity and increases the formation of ROS in the neuroepithelium, leading to leaky vessels, microhemorrhages, and worsening cognitive abilities.3-5 In addition, thickening of the neuroepithelial basement membrane further disrupts the blood-brain barrier, worsening neuroinflammation via the production of proinflammatory cytokines.4,18
To make matters worse, a state of brain hypoperfusion promotes protein aggregation and activates immune response via glial cells.5 If these changes progress, pathological entities, such as amyloid deposits and microhemorrhages, increase the disease burden.19 Furthermore, common comorbidities in AD, such as diabetes and hypertension, worsen this process through increased blood pressure, decreased cerebrovascular reactivity, and leaky blood vessels.5 Hypoperfusion and concurrent accumulation of amyloid correlates with increased cerebrovascular injury and brain atrophy. Therefore, metabolic derangements are a culprit of stepwise pathological processes of vascular and blood-brain barrier abnormalities that could be prevented with control of diabetes and hyperinsulinemia.12
Metabolic and mitochondrial defects have been studied in brains affected by AD. Studies have shown that patients with AD have a higher glucose concentration in the brain, leading to decreased cellular glucose use and concurrent insulin resistance.6 In addition, patients with AD are observed to have a higher fasting glucose compared to controls.6
A pathological cascade of metabolic defects shows a limitation to mitochondrial function via hyperglycemia and hyperlipidemia, leading to insulin resistance and the deposition of extracellular amyloid. Positron emission tomography (PET) scans of post-mortem AD brains reveal defects in insulin receptors localized to the blood-brain barrier, adding to an already altered insulin signaling, affecting glucose metabolism.6 Alterations in mitochondrial function lead to the disastrous consequences of increased susceptibility to cell death and neural senescence. Because of a decreased capability of glucose usage by damaged mitochondria, alterations of basal glucose levels by decreased ATP production in the brain cause episodes of hypoperfusion, leading to synaptic loss.12,18 Therefore, damaged mitochondria produce more ROS that damage surrounding tissues, becoming a vicious cycle of inflammation, oxidative damage, and neuronal loss. These structural and mechanical alterations are responsible for neuronal senescence and cognitive loss in AD.
Part 3: The Relationship Between Metabolic and Mitochondrial Health
Defects in glucose processing and insulin resistance are an early event in the development of cognitive impairment. Accumulation of amyloid with further destabilization of the blood-brain barrier causes stepwise vascular damage that worsens cognitive loss. Insulin resistance is shown to damage neuronal mitochondria by decreasing their ATP-producing capacities and increasing oxidative stress, which, in turn, contributes to neuronal senescence and synaptic loss.6,12,18
Damaged mitochondria accumulate in the neuron, worsening neuronal transmission of chemical messengers as well as destabilizing synaptic connections.6,12
Oxidative stress and production of ROS by dysfunctional mitochondria causes cell death and neuronal senescence. Oxidative stress activates cytochrome C, triggering the apoptotic pathway.19 On the other hand, a senescent neuron forms from continuous ROS production, leading to autophagic defect from lysosomes and the accumulation of damaged mitochondria and intracellular debris, such as iron deposits, that concurrently damage nucleotide formation and deoxyribonucleic acid (DNA).13,19,20
Studies also have shown that insulin resistance is related to the development of intracellular neurofibrillary tangles in patients with AD.12 In a normal brain, the role of tau protein is to regulate intracellular communication through the stabilization of neuronal microtubules. It is suggested that insulin resistance alters multiple protein kinases that are involved in tau protein hyperphosphorylation as seen in the development of tau neurofibrillary tangles.12 In addition, neuroinflammation, via proinflammatory cytokines such as interleukin (IL) 6 and tumor necrosis factor (TNF)-alpha, increases the formation of tau neurofibrillary tangles and contributes to the progression of AD through the destabilization of the synapse and neural death, specifically in the hippocampus.12,18
Like T2DM, the formation of AGEs in the brain also occurs in AD. The AGE molecules in amyloid plaques, because of hyperglycemia and insulin resistance, have been isolated in postmortem AD brains.12 Moreover, AGEs have been shown to be neurotoxic by increasing the production of ROS and promoting the formation of intracellular tau neurofibrillary tangles.12
Part 4: Treatments Targeted Toward Mitochondria Repair and Recovery
Mitochondrial dysfunction is a primary event in the pathogenesis of both AD and T2DM. Therefore, treatment modalities targeted toward mitochondria should be employed to decrease disease burden. (See Figure 4.) Studies show that mitochondrial function normalizes with exercise as it decreases tissue insulin resistance.6,19
Figure 4. Multifaceted Alzheimer’s Disease Treatment |
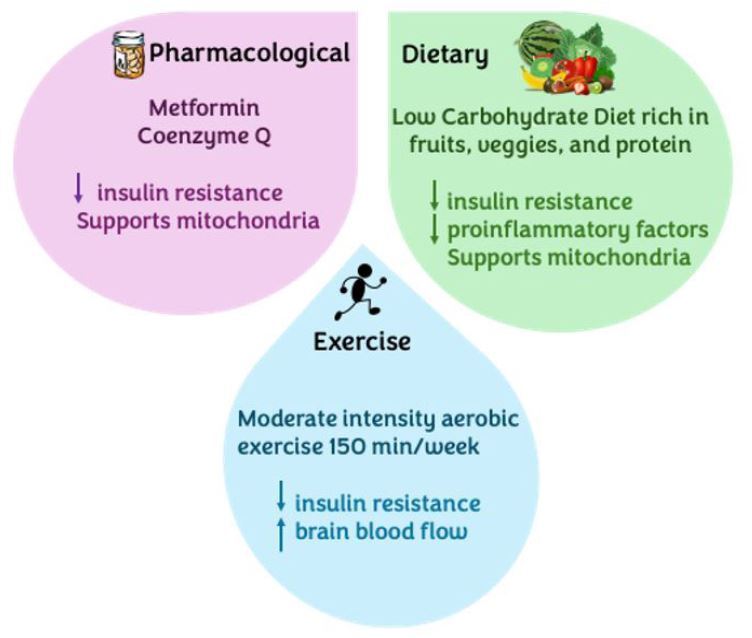 |
Created by Gabriela De Los Santos, MD. |
Ample research has shown how exercise increases blood flow and improves cognitive function, and even increases temporal lobe brain mass in Alzheimer’s patients.6,19 Exercise serves as a strong therapeutic and preventive measure when tackling Alzheimer’s and other dementias. Multiple studies highlight that the risk of developing dementia can be lowered by one-quarter.20
To be more specific, moderate-intensity aerobic exercise is extremely beneficial, improving memory, attention, and executive function in elderly patients who exercise compared to those with a sedentary lifestyle.20 Furthermore, the benefits of exercise can be directly observed through the preservation or even enlargement of the hippocampus, increased blood flow and oxygen delivery to brain areas (especially the anterior cingulate gyrus), decreased inflammation, and even a reduction in cortical decay.20
The most widely studied exercise modality that shows these benefits involves performing at least 150 minutes per week of moderately intense aerobic exercise.20 However, it is important to note that these studies imply a risk reduction in the development of cognitive disorders. In patients already diagnosed with AD, exercise as therapy has shown a reduction in neuropsychiatric symptoms and a slower progression of cognitive deterioration, not necessarily an improvement in memory abilities. Therefore, it is extremely important to place an emphasis on the prevention of AD before developing permanent structural brain deficits.
Multiple drugs, such as metformin, Szeto-Schiller 31 peptide, coenzyme Q, and imeglimin, have gained popularity as mitochondrial-targeting drugs in the treatment of AD. Several studies have elucidated the role of metformin specifically in the brain.21 In vitro studies have reported that metformin is able to suppress neural apoptosis induced by amyloid aggregates.22
It is important to note, though, that results are controversial, and other studies have shown the opposite effect. It is thought that metformin induces the formation of synapses and helps activate the process of spatial memory development by increasing the expression of several neural growth factors, such as brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF).22 Additionally, multiple animal studies have proposed the role that metformin plays in decreasing the aggregation of amyloid in the brain. It is hypothesized that metformin increases the levels of insulin-degrading enzyme, a protease known to cleave insulin and misfolded amyloid protein.22 Not only does this show a stronger relationship between AD and T2DM, but it also suggests a possible neuroprotective effect. However, it is noteworthy that some studies have shown an increase in amyloid toxicity with metformin use. Moreover, metformin repeatedly has been shown to decrease proinflammatory factors such as TNF-alpha, IL-1-beta, and IL-6, thus decreasing neuroinflammation.22
In terms of neurometabolic effects, metformin has been shown to decrease the risk of developing dementia by 35% in patients with T2DM.22 The mechanism is thought to be by decreasing insulin resistance through the upregulation of insulin-related tyrosine kinases, thereby increasing post-receptor insulin signaling and preventing further metabolic derangements.3,6,21
Metformin dosage has been studied limitedly in patients with mild cognitive impairment. A dosage of 1,000 mg twice per day for 12 months was shown to have better results in a selective recalling test compared to the placebo group.22 Larger studies need to be conducted to further elucidate the positive effects on cognitive abilities. Long-term metformin use is known to decrease vitamin B12 levels and can cause bothersome gastrointestinal side effects in some patients, which need to be monitored by a licensed provider.
Coenzyme Q has been observed to modulate insulin pathways and help in the replacement and replenishment of damaged mitochondria.3,6,23 Coenzyme Q is known to have antioxidant properties that serve to eliminate free radicals and even help with the generation of other antioxidants.23,24 Unfortunately, as mitochondria become damaged in AD, there is a release of ROS and other proinflammatory factors that amplify the inflammation cascade in the brain. Coenzyme Q is found especially in the inner mitochondrial membrane, where it works with the electron transport chain to generate ATP in the cell.24,25
Multiple studies have evaluated the use of coenzyme Q in patients with diagnosed AD. In most studies that showed positive changes, a daily dose of 200 mg was used.24 An increase in cerebrospinal fluid coenzyme Q was observed in patients taking their daily coenzyme Q dosage, indicating cerebrovascular absorption of this antioxidant.24
Other studies also reported the presence of this compound in amyloid and tau aggregates of AD patients.24,25 Not only was the antioxidant observed to be absorbed through the blood-brain barrier, but it also has shown positive results related to memory in patients with dementia.
In patients with diagnosed dementia, the administration of 200 mg of coenzyme Q per day showed improvements in neuropsychological tests.24 It also is noteworthy that in patients with vascular dementia, multiple studies revealed that the administration of coenzyme Q for about three months not only normalized measured serotonin levels in the cerebrospinal fluid (CSF) but also decreased the presence of certain inflammatory markers such as IL-6, TNF-alpha, and IL-1-beta.24,25 Even in patients with mild cognitive decline, the use of coenzyme Q (200 mg/day) was shown to improve cerebral vasoreactivity and cognitive function through certain neuropsychological tests.24
It is important for a licensed provider to monitor for reported side effects, such as nausea, vomiting, rash, and/or insomnia. There also have been some reports of drug interactions between coenzyme Q and warfarin.
Szeto-Schiller 31 peptide works by inhibiting oxidative stress in the mitochondria, thereby helping to restore their normal function.9,6,23 Szeto-Schiller 31 peptide is a mitochondria-targeting peptide that is theorized to inhibit peroxidation, thus decreasing the amount of reactive oxygen species produced by damaged mitochondria and aiding in the ability of the mitochondria to properly produce ATP after hypoxic damage.26 Most studies done on this novel antioxidant have been with mouse models; therefore, a set dose of this peptide has not been studied in human patients for T2DM and AD. However, this supplement does exist as a 10 mg daily tablet. Szeto-Schiller 31 peptide has been shown to cross the blood-brain barrier in in vivo studies.23 Furthermore, it was shown to both ameliorate mitochondrial function and decrease the amount of aggregated amyloid protein in the synapses of mouse models.23 Given that mitochondrial dysfunction is an early event in the pathogenesis of AD, further study of this peptide is warranted.
Imeglimin works by reducing ROS and enhancing the release of insulin from pancreatic beta cells.6 It has the mechanism of decreasing insulin resistance and production of ROS, leading to better glucose processing and less neurodegeneration.3,6,21,23
Although not currently approved by the U.S. Food and Drug Administration, imeglimin has gained popularity because of its anti-inflammatory and mitochondrial-targeting properties. For example, imeglimin has been shown to increase ATP generation, decrease reactive oxygen species formation, and decrease the release of pro-apoptotic proteins from the mitochondria.6,27-29 In addition, this new drug has antioxidant properties and is able to decrease insulin resistance by enhancing insulin signaling and glucose uptake into the cells.6,29 Multiple studies with human subjects have been done in Japan to further elucidate imeglimin’s treatment properties in T2DM and mild cognitive decline, and it was cleared for clinical use in patients with T2DM in 2021.6,27-29 With its ability to improve mitochondrial function and decrease insulin resistance, it is hypothesized that imeglimin may be beneficial for use in patients with cognitive decline.6,28 In the United States, imeglimin currently is in Phase II of clinical trials.
Brain inflammation is a key event in the progression of AD. Certain proinflammatory markers, such as IL-6, TNF-alpha, and IL-1-beta, have been implicated in the potentiation of the inflammatory cascade in the Alzheimer’s brain. Although it is not clinical practice to measure these inflammatory markers to track disease progression, certain studies have revealed that as a patient with AD worsens (as measured by neuropsychiatric symptoms and cognitive decline), the levels of these proinflammatory markers continue to increase, showing a positive correlation.30
Although biomarkers such as tau and amyloid are used to measure the efficacy of certain drugs in AD treatment, measuring proinflammatory markers also should be considered in this testing process. As a result, providers might be able to track disease progression using these proinflammatory markers.31
As discussed previously, T2DM is a strong risk factor for the development of AD and other dementias. Recent data have elucidated that tighter glycemic control may decrease a patient’s risk of developing dementia. Although the American Geriatrics Society recommends an HbA1c goal of 7.5 to 8, multiple studies have shown a decreased risk of cognitive decline with tighter glycemic control.32,33
In a study with about 55,000 patients with T2DM, results showed that having a glycemic control target of HbA1c between 6.5 and 7.5 may decrease dementia incidence by 28%.32 Therefore, a multidisciplinary approach in the primary care setting when treating metabolic disorders such as T2DM will indeed help in the prevention of cognitive dysfunction.
A ketogenic diet or therapeutic carbohydrate restriction in AD patients may decrease the progression of the disease.18 As previously discussed, the pathogenesis of AD is plagued with widespread neuroinflammation. The ketogenic diet has been shown to decrease proinflammatory cytokines such as IL-6 and TNF-alpha while increasing neuroprotective mediators such as neurotrophin-3, brain-derived neurotrophic factor, and glial cell line-derived neurotrophic factor.18,34 Therapeutic carbohydrate restriction also restores mitochondrial function through the inhibition of glycolysis by ketone bodies and a decrease in the production of ROS.12,18,34 This decrease in ROS promotes neuroprotection and the stabilization of DNA.
While there is controversy about how low-carbohydrate nutrition regimens may be associated with dyslipidemia, the clinical significance is unknown. Yet, by restricting carbohydrates, insulin resistance improves, thereby reducing the associated inflammatory pathways described earlier.3,10,13,14,18,19 Finally, therapeutic carbohydrate restriction is a modality that promotes the regulation of insulin levels that can be used to prevent and treat patients with metabolic derangements such as AD and T2DM.3,10,12-14,17,18
For example, the MIND (Mediterranean-DASH Intervention for Neurodegenerative Delay) diet uses a combination of the Mediterranean and Dietary Approaches to Stop Hypertension (DASH) diets to lessen the progression of cognitive decline. This diet limits carbohydrate intake to 130 net carbohydrates per day and is rich in vegetables, berries, nuts, omega-3-rich whole grains, fish, and poultry.35 A limitation also is placed on fried foods, refined sugars, margarine, and red meat because of their pro-inflammatory effects on the body.35
A small study of 31 patients with AD using the MIND diet found a significantly thicker cortex compared to patients who did not use this diet.35 This effect is thought to be mitigated by an amelioration of insulin resistance, reduction of neuroinflammation, and greater glycemic control, thus serving as a neuroprotective approach in the metabolically deranged brain of an AD patient.
Conclusion
Insulin resistance manifests itself in the muscle through the development of T2DM and in the brain through the development of AD. Damage in neural mitochondria leads to apoptosis and senescence, widespread neuroinflammation, and production of ROS that contributes to the progression of AD. Therefore, treatment modalities should be targeted toward the normalization of metabolic function. These include drugs that decrease oxidative stress in the mitochondria and regulate its function, drugs that mediate insulin resistance, and lifestyle modification, such as increased exercise and therapeutic carbohydrate restriction, to prevent and decrease the progression of AD.
References
- Goyal R, Singhal M, Jialal I. Type 2 Diabetes. StatPearls Publishing; 2024 Jan-. https://www.ncbi.nlm.nih.gov/books/NBK513253/
- Li X, Feng X, Sun X, et al. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990-2019. Front Aging Neurosci 2022;14:937486.
- Raut S, Bhalerao A, Powers M, et al. Hypometabolism, Alzheimer’s disease, and possible therapeutic targets: An overview. Cells 2023;12:2019.
- Jackson K, Barisone GA, Diaz E, et al. Amylin deposition in the brain: A second amyloid in Alzheimer disease? Ann Neurol 2013;74:517-526.
- Raz L, Knoefel J, Bhaskar K. The neuropathology and cerebrovascular mechanisms of dementia. J Cereb Blood Flow Metab 2016;36:172-186.
- Carvalho C, Moreira PI. Metabolic defects shared by Alzheimer’s disease and diabetes: A focus on mitochondria. Curr Opin Neurobiol 2023;79:102694.
- Xia X, Jiang Q, McDermott J, Han JDJ. Aging and Alzheimer’s disease: Comparison and associations from molecular to system level. Aging Cell 2018;17:e12802.
- Kumar A, Sidhu J, Forshing L, Tsao JW. Alzheimer Disease. StatPearls Publishing; 2024 Jan-. https://www.ncbi.nlm.nih.gov/books/NBK499922/
- Barbagallo M, Dominguez LJ. Type 2 diabetes mellitus and Alzheimer’s disease. World J Diabetes 2014;5:889-893. https://doi.org/10.4239/wjd.v5.i6.889
- Boden G. Effects of free fatty acids (FFA) on glucose metabolism: Significance for insulin resistance and type 2 diabetes. Exp Clin Endocrinol Diabetes 2003;111:121-124.
- Pinti MV, Fink GK, Hathaway QA, et al. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am J Physiol Endocrinol Metab 2019;316:E268-E285.
- Michailidis M, Moraitou D, Tata DA, et al. Alzheimer’s disease as type 3 diabetes: Common pathophysiological mechanisms between Alzheimer’s disease and type 2 diabetes. Int J Mol Sci 2022;23:2687.
- Kim J, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circulation Research 2008;102:401-414. https://doi.org/10.1161/circresaha.107.165472
- Singh VP, Bali A, Singh N, Singh Jaggi A. Advanced glycation end products and diabetic complications. The Korean Journal of Physiology & Pharmacology 2014;18:1-14. https://doi.org/10.4196/kjpp.2014.18.1.1
- Marzban L, Park K, Verchere CB. Islet amyloid polypeptide and type 2 diabetes. Exp Gerontol 2003;38:347-351. https://doi.org/10.1016/s0531-5565(03)00004-4
- Ebeling P, Koistinen HA, Koivisto VA. Insulin-independent glucose transport regulates insulin sensitivity. FEBS Lett 1998;436:301-303.
- Szablewski L. Brain glucose transporters: Role in pathogenesis and potential targets for the treatment of Alzheimer’s disease. Int J Mol Sci 2021;22:8142.
- Rusek M, Pluta R, Ułamek-Kozioł M, Czuczwar SJ. Ketogenic diet in Alzheimer’s disease. Int J Mol Sci 2019;20:3892.
- Ngandu T, Lehtisalo J, Solomon A, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): A randomised controlled trial. Lancet 2015;385:2255-2263.
- Meng Q, Lin MS, Tzeng IS. Relationship between exercise and Alzheimer’s disease: A narrative literature review. Front Neurosci 2020;14:131.
- Wiernsperger NF, Bailey CJ. The antihyperglycaemic effect of metformin: Therapeutic and cellular mechanisms. Drugs 1999;58(Suppl 1):31-39.
- Liao W, Xu J, Li B, et al. Deciphering the roles of metformin in Alzheimer’s disease: A snapshot. Front Pharmacol 2022;12:728315.
- Ding XW, Robinson M, Li R, et al. Mitochondrial dysfunction and beneficial effects of mitochondria-targeted small peptide SS-31 in diabetes mellitus and Alzheimer’s disease. Pharmacol Res 2021;171:105783.
- Jiménez-Jiménez FJ, Alonso-Navarro H, García-Martín E, Agundez JAG. Coenzyme Q10 and dementia: A systematic review. Antioxidants (Basel) 2023;12;533.
- Hidalgo-Gutiérrez A, González-García P, Díaz-Casado ME, et al. Metabolic targets of coenzyme Q10 in mitochondria. Antioxidants (Basel) 2021;10:520.
- Birk AV, Liu S, Soong Y, et al. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol 2013;24:1250-1261.
- Bando H, Yamashita H, Kato Y, et al. Combined treatment of vildagliptin/metformin (Equmet) and imeglimin (Twymeeg) with clinical efficacy. Asp Biomed Clin Case Rep 2023;6:69-75. https://asploro.com/combined-treatment-of-vildagliptin-metformin-equmet-and-imeglimin-twymeeg-with-clinical-efficacy/
- Bando H, Okada M, Iwatsuki N, et al. Improved HbA1c value by combined treatment of dulaglutide and imeglimin for patient with type 2 diabetes mellitus (T2DM). Int J Endocrinol Diabetes 2022;5:1-4. https://www.pubtexto.com/pdf/?improved-hba1c-value-by-combined-treatment-of-dulaglutide-and-imeglimin-for-patient-with-type-2-diabetes-mellitus-t2dm
- Dubourg J, Fouqueray P, Quinslot D, et al. Long-term safety and efficacy of imeglimin as monotherapy or in combination with existing antidiabetic agents in Japanese patients with type 2 diabetes (TIMES 2): A 52-week, open-label, multicentre phase 3 trial. Diabetes Obes Metab 2022;24:609-619.
- Mekli K, Lophatananon A, Maharani A, et al. Association between an inflammatory biomarker score and future dementia diagnosis in the population-based UK Biobank cohort of 500,000 people. PLoS One 2023;18:e0288045.
- Cummings J. The role of biomarkers in Alzheimer’s disease drug development. In: Guest PC, ed. Advances in Experimental Medicine and Biology. Springer Cham 2019;29-61. https://doi.org/10.1007/978-3-030-05542-4_2
- Wang K, Zhao S, Kam-Pui Lee E, et al. Risk of dementia among patients with diabetes in a multidisciplinary, primary care management program. JAMA Netw Open 2024;7:e2355733.
- Ramirez A, Wolfsgruber S, Lange C, et al. Elevated HbA1c is associated with increased risk of incident dementia in primary care patients. J Alzheimers Dis 2015;44:1203-1212.
- Broom GM, Shaw IC, Rucklidge JJ. The ketogenic diet as a potential treatment and prevention strategy for Alzheimer’s disease. Nutrition 2019;60:118-121.
- Bramen JE, Siddarth P, Popa ES, et al. Impact of eating a carbohydrate-restricted diet on cortical atrophy in a cross-section of amyloid positive patients with Alzheimer’s disease: A small sample study. J Alzheimers Dis 2023;96:329-342.
Diabetes is a strong risk factor for the development of Alzheimer's disease (AD) given the mitochondrial, vascular, and inflammatory modalities that contribute to its progression. Therefore, it is vital to understand the pathophysiology of these two diseases as they relate to insulin resistance and mitochondrial dysfunction. Exposing a metabolic relationship between these diseases can provide further understanding of metabolic health and how treatment of insulin resistance can decrease disease burden.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.