Metabolic Emergencies
Authors
Judith Toski Welsh, MD, Emergency Services Institute, Cleveland Clinic, Cleveland, OH
Purva Grover, MD, FACEP, Emergency Services Institute, Cleveland Clinic, Cleveland, OH
Core Content Outline: Inborn Errors of Metabolism
Urea Cycle Defects
Recognize the initial signs and symptoms of urea cycle defects
Plan the management of acute life-threatening processes resulting from urea cycle defects
Understand the pathophysiology of urea cycle deficiencies
Organic aciduria
Recognize signs and symptoms of clinical conditions characterized by the inherited organic aciduria disorders
Plan the initial management of acute life-threatening processes resulting from the inherited organic aciduria disorders
Understand the pathophysiology of the inherited organic acidurias
Galactosemia
Plan the initial management of the acute manifestations of galactosemia
Recognize the acute signs and symptoms of galactosemia
Understand the pathophysiology of galactosemia
Other
Understand the pathophysiology of other metabolic emergencies
Recognize the signs and symptoms of other metabolic emergencies
Glycogen Storage Disorders
Understand the general pathophysiology of glycogen storage disorders
Recognize the general signs and symptoms of glycogen storage disorders
Plan the initial management of a patient with the acute manifestations of a glycogen storage disorder
Urea Cycle Defects
Pathophysiology of Urea Cycle Disorders
Urea cycle disorders (UCDs) are genetic diseases causing a deficiency of one of the enzymes in the pathway for urea excretion. These are inherited in either an X-linked (ornithine transcarbamylase deficiency) or autosomal recessive manner (all the rest of the UCD). The urea cycle converts nitrogen from the metabolism of muscle and protein into urea (see Figure 1). Urea, unlike nitrogen, can be excreted because it is water soluble. Severe deficiency or total absence of activity of any of the first four enzymes (CPS1, OTC, ASS, ASL) in the urea cycle or the cofactor producer (NAGS) leads to the accumulation of ammonia and other precursor metabolites during the first few days of life. The brain is particularly sensitive to the toxic effects of ammonia. Seizures caused by elevated ammonia levels can also result in damage to the brain.
Figure 1
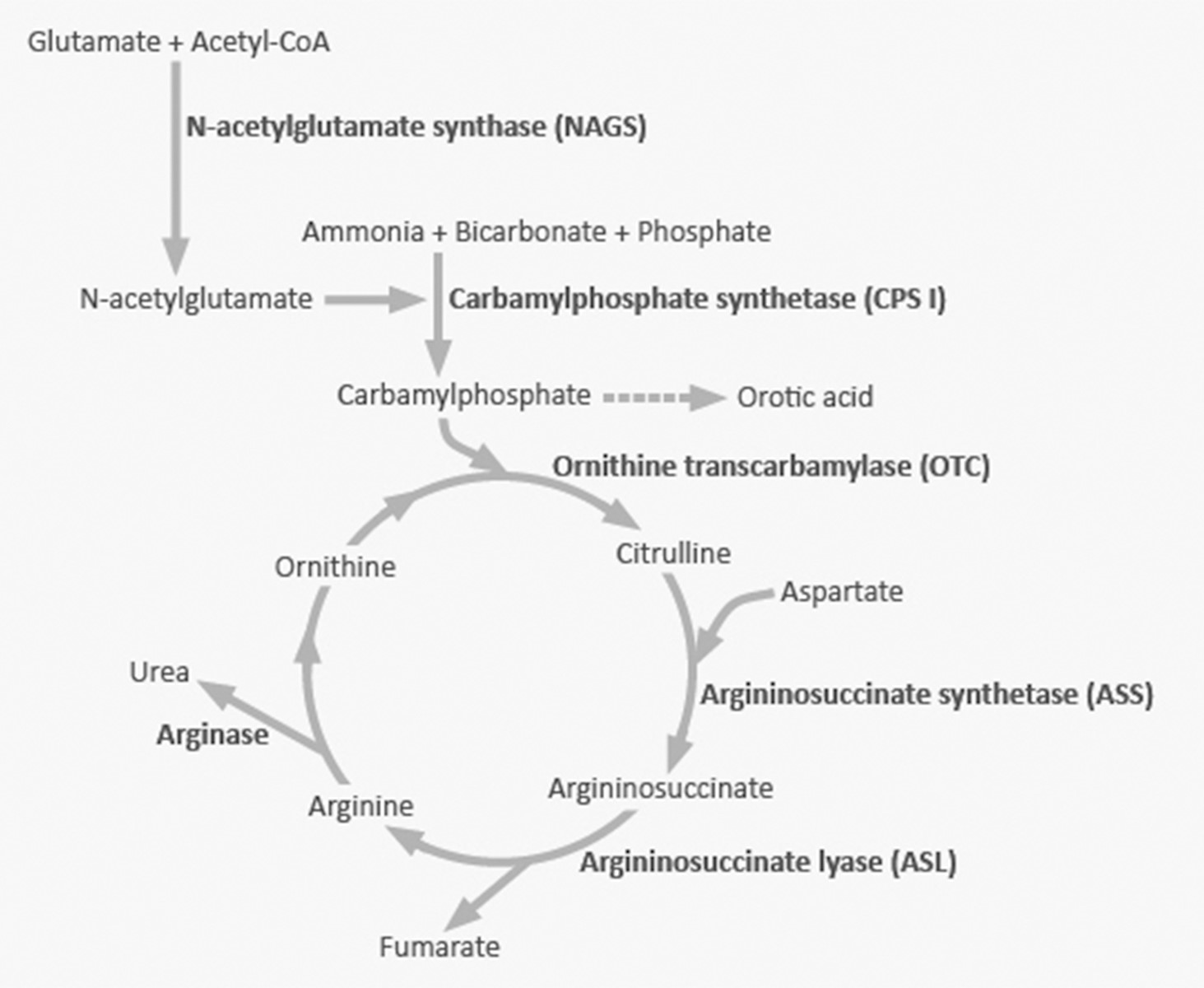
Signs and Symptoms
Neurologic and gastrointestinal symptoms are common and include altered mental status, abnormal motor function, seizures, vomiting, poor feeding, constipation, or diarrhea. Most patients develop symptoms in the newborn period or early childhood, but some patients with partial enzyme deficiency may present as older children or even in adulthood.
The typical patient is the neonate who appears well for the first 48 hours of life and subsequently develops poor feeding and vomiting, which progresses to lethargy and coma. Severely elevated ammonia levels lead to hyperventilation in newborns as a result of cerebral edema from the toxic metabolites. Other signs may include listlessness and poor tone. As it progresses, brain swelling can lead to hypoventilation and respiratory failure. Older children may demonstrate ataxia, developmental delays, psychiatric illness, chronic vomiting, or complain of chronic headache.
|
Symptoms of Urea Cycle Disorders In Newborns and Infants |
|
|
Symptoms of Urea Cycle Disorders in Older Children |
|
Management of Life-Threatening Presentations
The first step is to suspect a UCD and immediately assess and correct any abnormalities of the ABCs. Evaluating and securing the airway, ensuring adequate ventilation, and establishing IV access for volume expansion in the dehydrated child are the primary goals of stabilization.
Screening labs in any child with a critical illness or potential metabolic defect should include glucose, electrolytes, BUN and creatinine, arterial blood gas, lactic acid, and a urinalysis for ketones. In addition, ammonia, liver function tests, uric acid, urine reducing substances, and a CBC should be obtained. A urine orotic acid and organic acids level, the results of which are usually not immediately available, should be sent to help establish the diagnosis. Molecular genetic testing and specific enzyme analysis are used to help confirm the specific deficit.
Results that support the initial diagnosis of a urea cycle disorder include:
- Elevated serum ammonia level (> 150 mmol/L)
- Normal blood glucose
- Normal anion gap
Significant hyperammonemia is life threatening and must be addressed emergently. Severely elevated levels are often the consequence of UCDs. Coordinating patient management with pediatric critical care is important since children with neurotoxic blood levels or evidence of encephalopathy will require emergent dialysis. Treatment for acute severe hyperammonemia includes dialysis and hemofiltration, correction of dehydration with intravenous fluids, intravenous arginine hydrochloride and nitrogen scavengers to allow an alternate pathway for excretion of excess nitrogen, protein restriction, and delivering calories as carbohydrates and fats. Long term management includes dietary restriction of protein, specialized formulas, and oral nitrogen-scavenging drugs. All patients presenting acutely require admission to the hospital and management by a physician specialized in metabolic disorders.
REFERENCES/RESOURCES
Suggested guidelines for the diagnosis and management of urea cycle disorders
http://www.ojrd.com/content/pdf/1750-1172-7-32.pdf
Urea Cycle Disorders Review
http://www.ncbi.nlm.nih.gov/books/NBK1217/
Organic Acidurias
Pathophysiology
Organic aciduria is the excretion of non-amino organic acids in the urine. Most organic acidurias are the consequence of deficient enzyme activity leading to abnormal amino acid breakdown of branched chain amino acids or lysine. The most common diseases in this class are methylmalonic acidemia, propionic acidemia, isovaleric acidemia, and maple syrup urine disease.
The organic acidurias are inherited in an autosomal recessive manner. Many of these diseases are found at increased rates in populations where consanguinity is common, such as the Old Order Amish and in Arab populations in Saudi Arabia. Molecular genetic testing is used to confirm the diagnosis.[i]
Signs and Symptoms
Neonates generally appear well at birth, then present within the first days of life with vomiting, hypotonia, seizures, and altered mental status secondary to metabolic encephalopathy. Older children may develop symptoms subacutely in the setting of physical or psychological stress or dietary noncompliance.
Lab tests often show dehydration, hypoglycemia, elevated ammonia levels, ketonuria, and an anion gap acidosis. Symptoms can be mistaken for sepsis, especially since the white blood cell count and platelet count can be abnormally low.[ii] Decompensation in patients with maple syrup urine disease may be associated with no lab abnormalities.
|
Signs and Symptoms of Organic Acidurias |
|
Evaluation of an infant or child with a suspected organic aciduria presentation includes:
- Measurement of pH with a blood gas
- Bicarbonate
- Ammonia
- Lactate
- Glucose and electrolytes
- Renal function – BUN and creatinine
- Ketones
- Liver function tests
|
Disorder |
Distinctive Features |
||
|
Ketosis |
Acidosis |
Other |
|
|
Maple syrup urine disease (MSUD) 1 |
X |
Maple syrup odor |
|
|
X |
X |
Neutropenia |
|
|
Methylmalonic acidemia (MMA) |
X |
X |
Neutropenia |
|
Rare |
Rare |
Vomiting, poor feeding, neurologic symptoms |
|
|
Isovaleric acidemia 1 |
X |
Sweaty feet odor |
|
|
Biotin-unresponsive 3-methylcrotonyl-CoA carboxylase deficiency |
X |
Hypoglycemia |
|
|
3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) lyase deficiency |
Reye syndrome, hypoglycemia |
||
|
Ketothiolase deficiency (mitochondrial acetoacetyl-CoA thiolase deficiency) |
X |
X |
Hypoglycemia |
|
Glutaricacidemia type I (GA I) |
Basal ganglia injury with movement disorder |
||
- In MSUD and isovaleric acidemia, distinctive odors in urine, sweat, and even the affected individual's room suggest the diagnosis.
- Propionic acidemia may present with isolated hyperammonemia early in its course.
Management of Acutely Decompensated Organic Acidurias
After initial stabilization of the patient, the next priority is to correct hypoglycemia and to prevent a catabolic state. Low osmolarity solutions are preferred for the smallest patients. A symptomatic patient with a glucose level < 45-50 mg/dL should receive 2 mL/kg of 10% dextrose, followed by a dextrose infusion at 10-12 mg/kg per minute. The infusion should be titrated to maintain euglycemia. Presence of persistent hypoglycemia, especially with ketosis, suggests the presence of an inborn error of metabolism.[iii] Dextrose 25% or 50% may be used in older children to correct hypoglycemia. Insulin may need to be administered to maintain normoglycemia. Insulin should be started at 0.05 IU/kg/hour and adjusted as needed to maintain glucose between 100-120 mg/dL.[iv]
Correction of the metabolic acidosis, electrolyte abnormalities, and dehydration should occur simultaneously with management of hypoglycemia. Intravenous therapy is necessary in most patients due to intractable vomiting. Protein is withheld for 24-48 hours and then reintroduced gradually. Parenteral nutrition may be required if feeding intolerance persists despite resuscitation. Patients with severely elevated ammonia levels may require dialysis. L-carnitine is given either intravenously or orally to improve toxic metabolite excretion.
The recommended diet is low protein from food sources and supplementation with an amino acid mixture that excludes the substance which the patient cannot effectively utilize.
REFERENCES/RESOURCES
The Organic Acidemias: An Overview
http://www.ncbi.nlm.nih.gov/books/NBK1134/
Galactosemia
Pathophysiology of Galactosemia
An autosomal recessive disorder, galactosemia results from a deficiency of one of three enzymes that break down the sugar galactose. As a result, a toxic metabolite accumulates, resulting in failure to thrive, kidney and liver dysfunction, and sepsis, especially from Escherichia coli. Classic galactosemia, the most common and severe form of the disease, is characterized by deficiency of Galactose-1-phosphate uridyl transferase (GALT). Partial activity of GALT presents with multiple variants.
Signs and Symptoms
Neonates with galactosemia will present within days of ingesting breast milk or milk-based formulas with failure to thrive, low blood sugar, feeding problems, jaundice, and abnormal excessive bleeding. Unless galactosemia is treated with lactose-free formula, death due to septic shock may occur. If a neonate survives but continues to ingest lactose, the child risks severe brain damage and liver failure.
The most common and severe type of disease is classic galactosemia. Affected infants present in the first few days of life with jaundice, vomiting, diarrhea, lethargy, hypotonia, and failure to thrive. Evidence of liver failure may come in the form of edema, ascites, altered mental status, and excessive bleeding or bruising. Restriction of dietary galactose is the mainstay of treatment, starting in the neonatal period.
Characteristics laboratory findings in a child with classic galactosemia include:[v]
- Liver Dysfunction: Elevated bilirubin, elevated AST and ALT, coagulopathy
- Hemolytic anemia
- Metabolic acidosis
- Reducing substances in urine (galactosuria)
|
Signs and Symptoms of Galactosemia |
|
|
Long-Term Complications of Galactosemia |
|
Management of Acutely Decompensated Galactosemia
Initial management of infants with galactosemia includes management of the airway, breathing, and circulation (ABCs), and treatment for suspected sepsis. Once stabilized, a child with acutely decompensated galactosemia requires a lactose-free diet. Breast milk, whey protein, and any food that includes a milk source is forbidden. Soy formulas (Isomil or Prosobee), elemental formulas (Alimentum, Pregestimil, and Nutramigen, Neocate) are all appropriate for infants with galactosemia.[vii]
REFERENCES/RESOURCES
Classic Galactosemia and Clinical Variant Galactosemia
http://www.ncbi.nlm.nih.gov/books/NBK1518/
Glycogen Storage Disorders
Pathophysiology of Glycogen Storage Disorders (GSD)
Glycogen is the storage form of glucose present in liver and muscle tissue, and serves as a buffer for carbohydrate energy requirements. It is formed when sugar is plentiful in the diet and is broken down when needed during times of calorie deficit or physical stress. Glycogen storage diseases are a consequence of inherited enzyme defects that result in the inappropriate glycogen synthesis or breakdown. Most are inherited as autosomal recessive defects except for GSD Type VI.
Signs and Symptoms of Glycogen Storage Disorders
Children with glycogen storage disorders invariably present with signs and symptoms associated with severe hypoglycemia. Patients can present with dozens of different variants, from the neonatal period to adulthood, depending on the specific deficit. The disease process should be considered in patients who present with unexplained hypoglycemia and ketosis, with or without liver enlargement. Muscle cramps, exercise intolerance, easy fatigability, and weakness are common complaints. Symptoms improve with eating or administration of glucose. The first signs of disease may present at birth or much later depending on the exact enzymatic defect.
Table: Glycogen Storage Disorders and characteristic findings
|
Disease/Common Name |
Enzymatic Defect |
Physical Findings |
Important Lab Abnormalities |
|
Type I von Gierke |
Glucose-6-Phosphatase |
Large liver Growth retardation |
Lactic acid increased Metabolic acidosis Hyperlipidemia Hyperuricemia Fasting hypoglycemia |
|
Type II Pompe |
Lysosomal disease |
Cardiomegaly Hypotonia |
|
|
Type III Forbes-Cori |
Glycogen debranching |
Skeletal muscle weakness hepatomegaly |
Elevated LFTs Fasting hypoglycemia |
|
Type IV Andersen |
Glycogen branching |
Cirrhosis No muscle weakness |
Elevated LFTs Coagulopathy in late disease |
|
Type V McArdle |
Muscle phosphorylase |
Muscle weakness with exercise |
Rhabdomyolysis |
|
Type VI Hers |
Liver phosphorylase |
Hepatomegaly |
Fasting hypoglycemia |
|
Type VII Tarui |
Phosphofructokinase |
Skeletal muscle weakness |
Rhabdomyolysis, renal failure due to myoglobinuria[viii] |
Management of Glycogen Storage Disorders
The cornerstone of acute treatment is the immediate correction of hypoglycemia with intravenous glucose. Chronic management includes prevention of hypoglycemia by providing frequent feedings. Prolonged, severe hypoglycemia leads to developmental and physical disabilities, seizures, and death if not adequately treated. Episodes of hypoglycemia lead to lactic acidosis, elevated triglycerides and uric acid on lab tests, and liver enlargement. Dietary management of GSD includes restriction of sucrose and lactose. Fruit, juice, and dairy are eliminated from the diet. Cornstarch is given to prevent or treat hypoglycemia as it is a source of slow, steadily released glucose. Mineral and vitamin supplementation is needed, as osteoporosis is a common long-term complication of such a restricted diet.[ix]
In forms of GSD with a risk of rhabdomyolysis (see Table) burgundy or cola-colored urine is a sign that the patient requires intravenous fluids and evaluation of renal function and creatine phosphokinase. Urinalysis shows a dipstick positive for blood without RBCs present on the microscopic analysis. Half of patients with rhabdomyolysis will lack these findings on urinalysis, so a normal urinalysis does not rule out the condition.[x] Hyperkalemia is the most significant threat to life in patients with rhabdomyolysis and is present in 10-40% of cases. Arrythmias can occur as a consequence of hyperkalemia, and any patient with this condition should have a cardiac monitor placed. Acute renal failure also occurs as a consequence of acute tubular necrosis from the excessive quantity of filtered myoglobin from injured muscle cells.[xi]
REFERENCES/RESOURCES
Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics
www.acmg.net/docs/ACMG_Practice_Guideline_for_GSD_Type_I_GIM_online_Nov2014.pdf
Other Metabolic Emergencies
Patients with a known inborn error of metabolism can arrive in the ED prior to significant hemodynamic decompensation with apparently mild symptoms. These patients will often provide a letter from their metabolic specialist with specific instructions on how to manage their condition, from the appropriate labs to order to treatment plans and goals. Involvement of the metabolic specialist in each step of the treatment and disposition plan is the key to optimal management of these patients.
Significant hyperammonemia is life threatening and must be addressed emergently. Severely elevated levels are often the consequence of urea cycle disorders. Coordinating patient management with pediatric critical care is important, since children with neurotoxic blood levels or evidence of encephalopathy will require emergent dialysis. Sodium benzoate (500 mg/kg/day) and sodium phenylbutyrate (600 mg/kg/day) can be administered to allow nitrogen excretion through alternative pathways.[xii]
Cofactor supplementation may be appropriate in the ED. Pyridoxine (vitamin B6) should be considered for management of seizures in neonates refractory to conventional treatment. A single intramuscular injection of pyridoxine may be therapeutic and diagnostic. Carnitine supplementation is often recommended by metabolic specialists as treatment for fatty acid oxidation defects to allow the mitochondria to clear toxic metabolites. Biotin can be used to treat organic acid defects.
Table: Metabolic Emergencies and Diagnostic Testing
|
Ammonia |
Elevated in urea cycle defect, organic acidemia, fatty acid oxidation defects |
|
Lactate |
Elevated in sepsis, hypoperfusion, organic acidemias |
|
Uric Acid |
Elevated in organic acidemia and Maple Syrup Urine Disease |
|
Liver Function |
Abnormal in galactosemia |
|
Urine Reducing Substances |
Tests for carbohydrate metabolites. If glucose is negative on UA, suggests that other carbohydrates are present. + in galactosemia and fructosemia |
|
CBC |
Neutropenia and thrombocytopenia with organic acidemias |
|
Management of Metabolic Emergencies in the ED |
|
1. Manage ABCs 2. Stop all protein intake 3. Provide hydration with glucose supplementation, correct metabolic acidosis 4. Eliminate toxic metabolites 5. Treat precipitating factors, including sepsis 6. Provide cofactors 7. Appropriate disposition and coordination of care with subspecialists |
|
Pitfalls |
|
1. Failure to consider and treat sepsis, which frequently coincides with metabolic decompensation 2. Failure to provide pyridoxine for intractable neonatal seizures 3. Failure to consult the patient’s metabolic specialist about treatment and disposition 4. Failure to consider hemodialysis for severe hyperammonemia 5. Assuming that there is no metabolic disease process because the newborn screen was “normal”. |
[i] Seashore M. The Organic Acidemias: An Overview. In Pagon RA, Adam MP, Ardigner HH, et al, eds. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015.
[ii] deBaulny HO, Saudubray JM. Branched chain amino acidurias. Sem Neonatology 2002;7:65-74.
[iii] Kumar P, Saini SS. An Update on Neonatal Hypoglycemia, Hypoglycemia – Causes and Occurrences, Prof Everlon Rigoberto (Ed.) InTech. Available at: http://cdn.intechopen.com/pdfs-wm/21466.pdf. Accessed 11/23/14.
[iv] Weiner DL. Metabolic Emergencies. In: Textbook of Pediatric Emergency Medicine, 5th ed. Fleisher GR, Ludwig S, Henretig FM, eds. Philadelphia: Lippincott, Williams and Wilkins; 2006: 1193.
[v] Walter JH, Collins JE, Leonard JV. Recommendations for the management of galactosaemia. UK Galactosaemia Steering Group. Arch Dis Child 1999;80:93-96.
[vi] Schweitzer-Krantz S. Early diagnosis of inherited metabolic disorders toward improving outcome: The controversial issue of galactosaemia. Eur J Pediatr 2003;162(Suppl 1):S50-S53.
[vii] Bosch A. Classic galactosemia: Dietary dilemmas. J Inherit Metab Dis 2011;34:257-260.
[viii] Kishnani PS, Austin SL, Abdenur JE, et al. Diagnosis and management of glycogen storage disease type I: Practice guideline of the American College of Medical Genetics and Genomics. Genetics in Medicine 2014; 1-29.
[ix] Kishnani PS, Austin SL, Arn P et al. Glycogen storage disease type II diagnosis and management guidelines. Genet Med 2010;12:446-463.
[x] Young SE, Miller MA, Docherty M. Urine dipstick testing to rule out rhabdomyolysis in patients with suspected heat injury. Am J Emerg Med 2009;27:875-877.
[xi] Vanholder R, Sever MS, Erek E, Lameire N. Rhabdomyolysis. J Am Soc Nephrol 2000;11:1553-1561.
[xii] deBaulny HO. Management and emergency treatments of neonates with a suspicion of inborn errors of metabolism. Sem Neonatol 2002;7:17-26.
Ready to Test?
Metabolic emergencies