Acute Cardiovascular Events in Infants
Acute Cardiovascular Events in Infants
Introduction
Emergency medicine physicians routinely deal with cardiac emergencies in adult patients but rarely encounter infants with critical cardiac conditions. While the infant's cardiac physiology can be very different from an adult's, the general principles of preload, afterload, contractility, and vascular resistance are the same.
Neonatal Circulation
In utero, oxygenation occurs in the placenta. Low oxygen concentration has a vasoconstrictive effect on the vessels in the lungs; a high PaO2 causes vasodilatation. Fetal alveoli are filled with fluid, not inspired oxygen. As a result, pulmonary vascular resistance is very high. High fetal pulmonary pressures cause high right-sided heart pressures. Left-sided fetal heart pressures are low due to the low resistance of the placenta. As a result, the fetus has unique hemodynamics left heart/systemic pressures are lower than right heart/pulmonary pressures. (See Figure 1.)
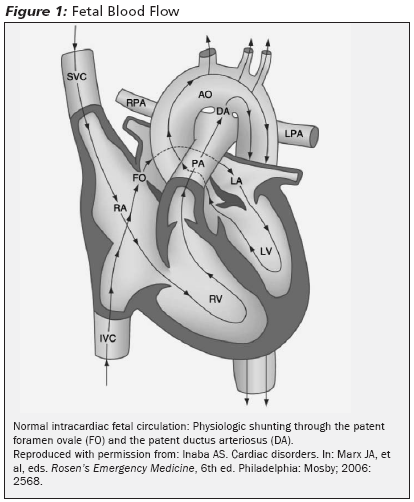 |
The foramen ovale and the ductus arteriosus are normal fetal structures that act as shunts. The direction in which fetal blood is shunted depends on the pressure difference between the pulmonary (right) and systemic (left) circulations. In the fetus, it is appropriate for blood to be shunted from the right to the left atrium through the foramen ovale, bypassing lungs that are not capable of oxygenation.1 The ductus arteriosus is a tubular connection between the pulmonary artery and the descending aorta. Because the systemic pressure is less than the pulmonary pressure in the normal fetal heart, blood is shunted from the pulmonary artery (right) to the aorta (left) through the ductus arteriosus, again bypassing the lungs.
Physiologic Changes at Birth
At birth, the left heart/systemic pressures are higher than right heart/pulmonary pressures. Fluid is compressed out of the lungs, and the alveoli are aerated. This increases the PaO2 and vasodilates the vessels in the lungs, decreasing pulmonary vascular resistance and right ventricular afterload. Twenty-four hours after birth, pulmonary pressures usually are reduced by half. The remainder of the drop in the pulmonary pressures occurs gradually over 2-6 weeks from birth.2,3 Clamping of the umbilical cord increases the newborn's systemic/left-sided heart pressures by increasing systemic vascular resistance. Once the right-sided heart pressure is less than the left-sided heart pressure, the foramen ovale closes.4,5 The increased PaO2 in the pulmonary artery signals the ductus arteriosus to contract.
Congenital Heart Disease
Epidemiology and Etiology. Congenital heart diseases (CHD) are the most frequently occurring birth defects, with an incidence of 4-8 per 1,000 live births.6-8 CHD accounts for 9% of deaths in infancy.7,9 A significant percentage of patients with CHD require surgery in the first year of life. Most deaths in CHD occur in the first year of life.10
CHD can occur from single gene mutations in an autosomal dominant, recessive or X-linked manner or from chromosomal abnormalities.11,12 Having a parent with CHD increases the risk of CHD in a child by 10%.13 When one child has CHD, siblings are three times more likely to be born with CHD.14 Tetratogens that can lead to CHD include ethanol, phenytoin, retinoic acid, and lithium. Certain maternal diseases are associated with CHD, such as diabetes, systemic lupus erythematosus, and phenylketonuria.
Prenatal cardiac screening by routine ultrasound done at 18-22 weeks typically detects 25-57% of CHD.15-18 Thirty-five percent to 45% of infants with CHD are detected in the newborn nursery.7,18,19 Of the infants with CHD who are undiagnosed in the nursery, 35% are diagnosed by 6 weeks of age and 57% by 3 months.19 These infants most often present with shock (not cyanosis), followed by murmur and respiratory distress.18
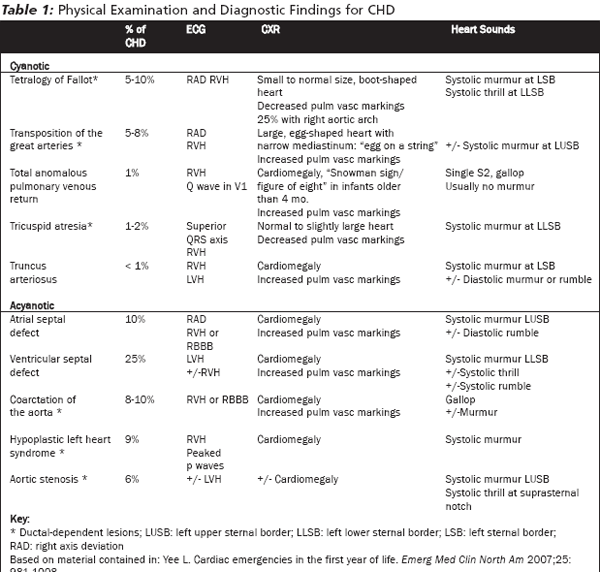
Congenital heart disease may be cyanotic or acyanotic. (See Table 1.) Acyanotic lesions include coarctation of the aorta and hypoplastic left heart the two lesions most likely to present as an infant in shock. Cyanotic lesions can be treated by attempting to improve blood flow to the lungs. CHD can be classified into three clinically relevant categories: ductal-dependent lesions, left-to-right shunt lesions, and total mixing lesions.
 |
Ductal-Dependent Lesions
Closure of the ductus arteriosus is a gradual process that occurs anywhere from 10 hours to 3 weeks after birth.20 The presence of the ductus arteriosus and the foramen ovale allows the fetus with abnormal cardiac anatomy to survive heart defects that will become symptomatic once physiology changes at birth. While the foramen ovale cannot be reopened in the ED, the ductus arteriosus can be chemically induced to remain open. Opening the ductus helps to reestablish the protective neonatal hemodynamic flow pattern until a more definitive repair is possible.
Infants with ductal-dependent lesions typically present in the first 3 weeks of life when the duct closes and present with shock or cyanosis. (See Figure 2.) Ductal-dependent lesions can be divided further based on how the ductus is utilized.
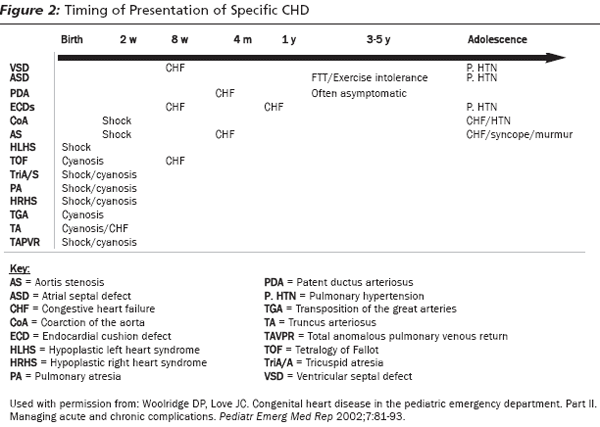 |
Ductus for Alternate Pulmonary Blood Flow. Many congenital heart defects obstruct normal blood flow from the right ventricle to the pulmonary artery and need the ductus to allow blood to reach the lungs. These include tetralogy of Fallot with severe right ventricular outflow obstruction (TOF), tricuspid atresia, transposition of the great arteries (TGA), hypoplastic right heart syndrome (HRHS), and pulmonary atresia. All of these lesions present with cyanosis as the duct closes. (See Figure 2.)
Tetralogy of Fallot is the most common form of cyanotic CHD.2 (See Table 1.) The abnormalities associated with TOF are a large ventricular septal defect (VSD), right ventricular outflow obstruction, right ventricular hypertrophy (RVH), and an overriding aorta. Those with severe right ventricular outflow tract obstruction will present in the first few weeks of life when the duct closes. Those with mild obstruction will present later with congestive heart failure (CHF) due to the long-term increased right-sided heart pressures, or with cyanotic spells ("tet spells"). (See Figure 2.)
Ductus for Alternate Systemic Blood Flow. Three lesions need the ductus to deliver blood to the systemic circulation: severe coarctation of the aorta (COA), critical aortic stenosis, and hypoplastic left heart syndrome (HLHS). In utero, these cause significant obstruction to normal blood flow, and the ductus is used for blood flow to the body. For example, in the fetus with coarctation of the aorta, a flange of tissue obstructs blood flow through the aorta. Blood flows from the pulmonary artery through the ductus to the aorta and the body. After birth, when the ductus closes, systemic blood flow is dramatically decreased. These lesions, therefore, typically present in the newborn period in the first 3 weeks of life with shock.20
HLHS is the most common deadly defect in the neonatal period and always presents with shock. It is fatal if untreated.21
Left-to-Right Shunt Lesions
This category includes ventricular septal defects (VSD), atrial septal defects (ASD), patent ductus arteriosus (PDA), and endocardial cushion defect. The various endocardial cushion defects lead to ASD, VSD, and mitral and tricuspid valve abnormalities. At birth, the hemodynamics shift, resulting in systemic/left ventricular pressures being higher than pulmonary/right ventricle pressures. Consequently, any defect in the interventricular septum, whether at the atrial or ventricular level, will cause an acyanotic shunt with blood flowing from the left to the right heart. The volume of blood that is shunted will determine the degree of right chamber enlargement and hypertrophy. The greater the flow, the more likely a murmur will be auscultated on examination. The amount of flow also dictates the timing and manner of presentation. Higher flows will cause earlier and more severe CHF presentation.22 (See Figure 2.) As shunt flow through the defect increases, the flow out of the right ventricle and into the pulmonary system increases. Infants with large left-to-right shunts can present in CHF.
Total Mixing Lesions with Abnormal Pulmonary Blood Flow
The two main mixing lesions are truncus arteriosus and total anomalous pulmonary venous return (TAPVR). Significant mixing of desaturated blood with the systemic-bound blood causes the cyanosis. Usually they are accompanied by an obstruction to pulmonary blood flow and, therefore, suboptimal oxygenation of the blood, which compounds the cyanosis. These two cyanotic CHD lesions are not ductal-dependent. Consequently, they do not respond to PGE1. Collectively, these represent less than 2% of CHD. (See Table 1.)
In the normally developing fetal heart, the truncus arteriosus eventually splits into the pulmonary artery and the aorta. Failure of this separation results in a single arterial trunk. If pulmonary obstruction is present, cyanosis and shock occur in the first few weeks of life.
In TAPVR, the pulmonary veins deliver blood to the right atrium. Blood travels from the right atrium to the left atrium through an ASD. If there is pulmonary outlet obstruction, patients present in cyanosis and shock.
Physical Examination
A simplified set of vitals for birth to 1 year are: heart rate 140, RR 40, and a minimum systolic blood pressure of 60 mmHg.23,24
Assess for cyanosis, pallor, respiratory distress, or an ashen complexion. Patients with ductal-dependent lesions who present in shock will have diminished pulses. Differences in timing and intensity of the left brachial artery and the femoral artery suggests COA.25 Auscultate the precordium for murmurs, clicks, or gallops. (See Table 1.) Palpate the precordium for thrills. A thrill may be felt at the suprasternal notch (AS), the left lower sternal border (VSD), or upper sternal border (pulmonary stenosis associated with several CHD defects). Murmurs louder than a grade 3 (moderate intensity), with an associated thrill, or hyperdynamic precordium are concerning. A diastolic murmur in an infant is never normal.25
Every effort should be made to keep the infant in a neutral temperature environment.26 Both low and high temperatures will cause metabolic stress, and thus cardiac stress, in the infant.
Diagnostics. Pulmonary markings are decreased in CHD lesions with low pulmonary blood flow. (See Table 1.) Look for a right aortic arch or cardiac malposition (dextrocardia), as these abnormalities can be seen in conjunction with CHD.27 CHD increases the risk of lower respiratory tract infections.5
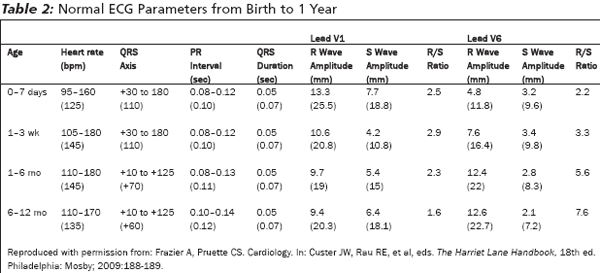
In the fetal heart, the majority of the blood flow is pumped by the right heart, which makes the right ventricle the larger, more muscular chamber at birth. As a result, there is a rightward axis deviation and RVH on ECG until 1-2 months of age.28 LVH is never normal in the first two months of life. As the infant grows, the left ventricle will grow, catch up, and exceed the size of the right ventricle. This dynamic leads to the progressively shifting axis seen throughout infancy. (See Table 2.)
 |
An arterial blood gas (ABG) is indicated in all cyanotic infants and in most infants who present in respiratory distress or shock.21 Complete blood count (CBC), chemistry panel, and calcium are indicated. Metabolic stress that occurs during hypotension or infection can cause significant hypoglycemia. Prolonged hypoglycemia can cause permanent brain damage in neonates and infants.29 Generally, a glucose level less than 40-45 mg/dL is considered low in this age group. However, if the infant's mental status is poor and blood glucose is 50-60 mg/dL, treat with glucose. Start at 4 mL/kg (0.4-0.5 grams/kg) of 10% dextrose.29
The Neonate Presenting with Cyanosis in the First 3 Weeks of Life
Most cyanotic CHD manifests in the first month of life.30 The vast majority of these are ductal-dependent and can be treated with PGE1. (See Table 1.) For the purpose of evaluation and stabilization, cyanosis can be divided in to central and peripheral forms.
Central cyanosis is caused by reduced arterial oxygen saturation. The tongue and mucous membranes appear blue. The main causes of central cyanosis are central nervous system (CNS), pulmonary, and cardiac.30 A significantly decreased PaO2 may exist without cyanosis.2
In peripheral cyanosis, the patient has a normal PaO2 but has increased extraction of oxygen.31 This is the result of slow movement of blood in the capillary system, allowing more time for oxygen to unload. This slow movement of blood can be due to vasomotor instability (neonatal acrocyanosis), vasoconstriction due to cold exposure, peripheral venous obstruction, polycythemia, shock, or methemoglobinemia. Peripheral cyanosis is visible in the hands, feet, and lips.
Nearly all (98%) of cyanotic cardiac lesions in the first 2-3 weeks of life will respond to Prostaglandin E1 (PGE1) because of their ductal dependence. PGE1 can be a life-saving treatment for these infants.
First, determine if the cyanosis is central or peripheral. If pulse oximetry is less than 90%, the cyanosis can be considered central. If the oxygen saturation is higher than 90%, a room air ABG can be helpful. The right radial artery is preferred, as it is preductal. A PaO2 less than 80 mmHg suggests central cyanosis.32,33 Fetal hemoglobin gives a falsely high reading on pulse oximetry.5,21
If the room air pulse oximetry reading is less than 85% and there is no increase in pulse oximetry with 10 minutes of administration of 100% oxygen, the cyanosis can be assumed to be cardiac, and PGE1 treatment should be started. If these conditions are not true, a formal hyperoxia test must be performed.1,2,26,34,35 In the hyperoxia test, an ABG is obtained after 10 minutes of 100% O2. A pulmonary-associated cyanosis will increase the PaO2 to greater than 150 mmHg (increase is usually greater than 30 mmHg from room air PaO2), while in cardiac-associated cyanosis, the PaO2 will not exceed 100 mmHg.33,34 Values between 100 mmHg and 150 mmHg are indeterminate, and the differentiation will need to be made by echocardiography.33
If the echo can be done quickly and the patient is stable, a hyperoxia test is not needed.2 If an ABG cannot be obtained, emergent echocardiogram is indicated.
If a cardiac cause of cyanosis is verified, start treatment with prostaglandin E1 (PGE1) as an IV infusion starting at 0.05 to 0.1 µg/kg/min.36 If there is no improvement in oxygen saturation after 30 minutes, increase the dose. Wait 20 minutes between dosage increases. If needed, the dose can be increased to a maximum rate of 0.4 µg/kg/min.30 When the effect is achieved, reduce the dose by increments of 0.01 µg/kg/min to the lowest infusion rate required to maintain the response.30 Wean supplemental oxygen as soon as possible.
Have age-specific airway equipment ready before administering the PGE1 since a common side effect is apnea (12%).21 PGE1-associated apnea is not dose related. Therefore, if the patient develops apnea, instead of decreasing an effective dose, intubate the patient.37 Apnea usually occurs within the first few hours of PGE1 treatment but can occur at any time during its administration.35 PGE1 may also cause hypotension, bradycardia, hyperpyrexia, and flushing. Hypotension is treated with a 10-20 cc/kg IV bolus of normal saline.35 If hyperpyrexia develops, administer acetaminophen rectally at a dose of 15 mg/kg.
If the maximum dose of PGE1 is reached without effect, the cyanosis may be due to non-ductal dependent lesions: truncus arteriosus or TAPVR. (See Table 1.) However, these cause less than 2% of cyanotic CHD. PGE1 is a life-saving treatment for the vast majority of patients. If acute worsening occurs on PGE1, stop the PGE1 and arrange for an emergent echocardiogram.37
The Neonate Presenting in Shock in the First 3 Weeks of Life
Some ductal dependent lesions do not present with cyanosis. (See Table 1.) When the ductus closes, systemic blood flow is dramatically decreased, leading to shock. Patients will present in the first two to three weeks of life with mottled skin and poor pulses. Sepsis is a more common cause of shock in the newborn period.
Clues to CHD cause for shock include cardiomegaly, pulmonary edema, or an ECG with hypertrophy. Murmurs are not always present in a patient who is in shock with CHD.38
Because of the potential for serious adverse effects, PGE1 should be given cautiously. It may worsen hypotension in both a septic or CHD patient and may cause apnea. However, giving PGE1 in ductal-dependent shock is the only life-saving treatment available for palliation until definitive correction can be accomplished surgically. Delay in administration will lead to prolonged shock and organ damage.
In an unstable infant in the first three weeks of life (when duct closure occurs), the presence of cyanosis or diminished pulses alone increases the probability of a correct diagnosis of CHD sufficiently to justify starting PGE1.39 This is supported by the recommendations of several authors.20,21,32,37,39 Therefore, the pediatric patient in the first three weeks of life presenting in shock with diminished pulses, even in the absence of a murmur, should receive PGE1.
If the shock is not adequately improved with PGE1, start dopamine or dobutamine.35 In conjunction with inotropic support, replace fluids, correct anemia, and confirm that PGE1 is at an adequate dose.40 Dopamine should be given through a central venous line starting at 2-5µg/kg/min and titrated up to 15 µg/kg/min.31,41 Dobutamine may be given peripherally if central access cannot be obtained. Start at 2.5-5 µg/kg/min and titrate to a maximum of 15 µg/kg/min.31,41 In the neonate in shock with normal or low heart rate, start epinephrine in a central venous line at a rate of 0.05-0.1 µg/kg/min and titrate to 1 µg/kg/min for improved perfusion.35,41
Transport
Once stabilized, transfer these patients to a center that offers pediatric cardiology and pediatric cardiac surgery subspecialty services. Strongly consider intubating any neonate on a PGE1 infusion before transport to protect the airway in the event of an apnea episode during transport.21,37 There is controversy as to whether it is necessary to intubate every infant being transferred on PGE1.42,43
Management of Acute Issues in the Infant with Known CHD
Complications of CHD and its corrective surgeries include conduit occlusion, endocarditis, arrhythmias, and tet spells.
The operative management of CHD often is staged and may involve prosthetic conduits or shunts. These conduits can become occluded with thrombus either acutely or gradually. Acute conduit obstruction will present to the emergency department with progressive cyanosis, low blood pressure, and/or cardiovascular collapse. Gradual obstruction presents with signs of right- or left-sided heart failure including hepatomegaly, peripheral edema, and jugular venous distention associated with superior vena cava syndrome. Heparin or thrombolysis may be indicated in consultation with a cardiologist.10
Many congenital heart defects are associated with a higher rate of dysrhythmias. This can be due to a congenitally abnormal conduction system or an irritable focus. Patients with CHD who have had operative repair may have scar formation that can serve as location for re-entrant tachycardias. The most common arrhythmias in the postoperative period are supraventricular tachycardia, ventricular tachycardia, sick sinus syndrome, and complete heart block.44
Tet spells are cyanotic episodes in Tetralogy of Fallot. TOF infants normally have left-to-right (acyanotic) shunting of blood across their VSD. But an increase in right-sided heart pressure caused by crying, feeding, or respiratory illness can cause reversal of the shunt. During tet spells, deoxygenated blood shunts from the right heart to the left heart, causing cyanosis. In addition to cyanosis, infants will present with rapid deep breathing, irritability, uncontrolled crying, and, on occasion, loss of consciousness. On auscultation, the cardiac murmur normally present will be decreased or absent.
Treat tet spells with supplemental oxygen, which may assist in dilating the pulmonary vasculature. Give morphine at 0.05-0.2 mg/kg/dose intramuscularly (IM), subcutaneously (SQ), or intravenously. Since crying worsens tet spells, it is better to place an IV to avoid multiple injections. Give a 20 cc/kg bolus of normal saline intravenously to improve preload and therefore right ventricular filling. Keep the infant as calm as possible. Allow the parent to hold the infant over his or her shoulder with the infant's knees bent to increase systemic vascular resistance and improve venous return to the heart.
If the symptoms do not resolve, give phenylephrine 5-20 µg/kg IV/IM every 10 minutes10 to increase systemic blood pressure and reverse the shunt.35 Propranolol can be given to improve symptoms at a dose of 0.01 to 0.2 mg/kg IV given over 5 minutes.4 Propranolol is thought to relax the right ventricular outflow obstruction. Rarely, all measures will fail and an emergent aorticopulmonary shunt will be needed.
Use an infusion filter such as a Pall IV filter when administering intravenous solutions to patients with known uncorrected septal defects. This filter prevents air bubbles in the IV line from crossing from the right to the left heart through septal defects and embolizing to the brain.
Congestive Heart Failure
Epidemiology and Etiology. Ninety percent of CHF seen in pediatrics occurs in the first year of life.36 (See Table 3.)
 |
History. Acute decompensation of CHF in infants is caused by exertion or illness (particularly pulmonary illnesses). Feeding has been described as a stress test for infants.4 Parents may report that the baby refuses to feed, is irritable with feeds, or feeds very slowly, often taking greater than 30 minutes per feeding.1 Prolonged feeding times often are accompanied by sweating or dyspnea. Sympathetic tone is increased, leading to cold, wet skin. CHF also may present non-acutely in infancy as failure to thrive.
Physical Examination. Tachycardia and a gallop rhythm frequently are present.31 Left-sided heart failure will present with rales or wheezing on pulmonary auscultation.30 Right-sided heart failure will present in infants with an enlarged liver and puffy eyelids. Infants may have biventricular failure and consequently have a combination of these findings. Jugular venous distention, peripheral edema, and anasarca are physical signs not commonly seen in infants with heart failure.3
Diagnostic Studies. Cardiomegaly on CXR may be present in both left- and right-sided heart failure. In infants, the thymus creates a shadow that can be difficult to distinguish from the heart border. If this presents difficulty during evaluation, obtain a lateral CXR. The retrocardiac space normally is a dark area the same size and lucency as the retrosternal space.45 In patients with cardiomegaly, the retrocardiac space will be replaced by the enlarged heart.3 Pulmonary vascular markings will be increased. Pulmonary effusions are not commonly found on CXR in infants with CHF.
ECG may reveal arrhythmias, hypertrophy, chamber dilation, or abnormal electrical axis. Laboratory studies should include a complete blood count and chemistries. Brain natriuretic peptide (BNP) levels are elevated in the first few weeks of life, but after the early neonatal period the BNP will be equivalent to adult normal levels.30,46,47
Management. If respiratory failure accompanies CHF, intubate the patient to support oxygenation and ventilation.23 If the infant is not in imminent respiratory failure, have him or her held upright to decrease pulmonary blood volume.4 Treat fever when it is present, as it represents increased metabolic demand on the heart. Give a dose of morphine (0.05-0.1 mg/kg/dose) to decrease agitation and air hunger.31 If there is increased work of breathing due to pulmonary edema, confirmed on CXR, give furosemide starting at a dose of 0.5 mg/kg/dose to 1 mg/kg/dose, with a maximum dose of 2 mg/kg/dose.41,46 Correct anemia to a hematocrit of greater than 35%.30 Slowly transfuse packed red blood cells 5-10 cc/kg over at least 4 hours.46
Start inotropic support in CHF patients presenting in cardiogenic shock. Refer to previous section for dosing information of inotropes. In the infant with decompensated CHF and adequate blood pressure, IV vasodilators may be used to reduce afterload and improve cardiac output.46 Nitroglycerin can be started at 0.25-0.5 µg/kg/min and titrated up by 0.5-1.0 µg/kg/min every 3-5 minutes, with a maximum dose of 20 µg/kg/min. The usual dose is 1-5 µg/kg/min.31,41
Milrinone is a phosphodiesterase-III inhibitor that can be given through a peripheral IV and has the advantage of having no chronotropic effects. Milrinone should be used in consultation with a pediatric cardiologist. Load with a dose of 50 µg/kg over 15 minutes followed by a maintenance infusion of 0.25-0.75 µg/kg/min, titrating to effect.41,46
Disposition. Newborns in congestive heart failure should be admitted to an intensive care unit, preferably a pediatric ICU, for stabilization and further evaluation. An echocardiogram will help confirm if a congenital heart defect is the cause of the CHF.
Arrhythmia
Epidemiology and Etiology. Supraventricular tachycardia (SVT) is the most common arrhythmia in infancy and childhood. Estimates of prevalence of SVT range from 1/250-1/1,000, with the higher prevalence arising from more contemporary studies.48,49 Fifty percent of SVT cases in infants are idiopathic, 20% are caused by CHD, and 20% by medications and other illnesses. Wolff-Parkinson-White syndrome (WPW) accounts for 10% of SVT in infants.50 Most cases of SVT in infancy present in the first 4 months of life, with a male predominance (male to female ratio of 3:2).51
History. Infants most commonly present with nonspecific symptoms such as irritability, feeding difficulty, cough, lethargy, or pallor as a result of their SVT. These symptoms have a recent onset, usually within 24 hours, but may be up to 48 hours. The symptoms represent some level of CHF in most patients. It is less common for infants to present in cardiogenic shock as a result of SVT.
Development of CHF is related to the duration of tachycardia. In one study, no patients with SVT for less than 24 hours developed CHF, while 20% of SVT for greater than 36 hours developed CHF. At greater than 48 hours, CHF occurred in 50%.49 Therefore, infants may be in SVT for days before presentation.
Diagnostics. ECG or rhythm strip will reveal a narrow-complex rhythm, usually with no discernable P waves, and no beat-to-beat variability. The heart rate in SVT in the infant is greater than 220 beats per minute (bpm) and may be as high as 280 bpm.28 Widened QRS in SVT secondary to aberrancy is rare in infants. If QRS aberrancy occurs, it is usually not sustained.52 CXR may reveal pulmonary edema.
Management. If the patient with SVT is in shock, perform immediate synchronized DC cardioversion at 0.5 to 1 J/kg. If this is ineffective, the voltage can be increased to 2 J/kg. A study attempting to determine the lowest effective energy for conversion of atrial dysrhythmias in the pediatric population found the successful shock to be 0.35 +/- 0.19 J/kg.53
If the patient is not in shock, first attempt vagal maneuvers. In an infant this is best accomplished by an ice bag or ice water to the perinasal area for 5-10 sec.31 This evokes the diving reflex. While carotid sinus massage and Valsalva maneuvers have been evaluated in older children, they are not feasible or effective in infants.54 There is one case report of cold fat necrosis resulting from application of ice to the face of a neonate in the setting of repetitive use.55 The authors recommend a short duration of ice exposure (5-10 sec.), with a cloth between the ice and the face, applied to the least fatty areas of the face. This maneuver will terminate SVT in 90-100% of patients.56 It is important to monitor for the cardioversion with a rhythm strip.57
If vagal maneuvers are ineffective, administer adenosine at 0.1 mg/kg with a maximum first dose of 6 mg.51 The half-life of adenosine is 10-15 seconds. If there is no effect with the first dose, there is no need to wait to administer the second dose. The second dose is 0.2 mg/kg, with a maximum dose of 12 mg.51 In one study, 68% of infants given adenosine were successfully cardioverted. The most effective dosing range was found to be 0.1-0.3 mg/kg.58 Adenosine may be given IV or by the intraosseous (IO) route.59 Note that patients who have previously undergone cardiac surgery may have a higher risk of side effects from adenosine. Therefore, use 70-80% of the recommended dose in these patients.58,60
If adenosine fails, consider synchronized electrical cardioversion or the addition of another antiarrhythmic. Consult cardiology. Amiodarone has been studied in the pediatric population with mixed results.61-63 The amiodarone is given at 5 mg/kg IV over 20-60 minutes with a maximum single dose of 150 mg.51 Reported side effects include hypotension, bradydysrhythmias, and heart blocks. Alternatively, give procainamide at a dose of 15 mg/kg IV over 30-60 minutes.51 Verapamil should not be used in infants for treatment of SVT due to reports of hypotension, bradycardia, and cardiovascular collapse.64
Digoxin and b-blockers are the first-line agents for maintenance therapy for infants with SVT. SVT recurs in approximately 30-33% of infants, although this rate may reach 44% later in infancy.65,66
Wolff-Parkinson-White (WPW) syndrome creates a dysrhythmia via an accessory pathway outside of the AV node.67 One study found that 34% of cases presented in the first 6 months of life. Thirty-one percent were associated with underlying CHD.68 (See Figure 3.)
 |
SVT in the setting of WPW usually occurs in the first year of life.31 Often, it isn't until the SVT has been converted to sinus rhythm that the ECG will reflect the characteristics of WPW. For this reason, it is important to get a 12-lead ECG after a patient with SVT converts to sinus rhythm to look for WPW.
Atrial fibrillation in the presence of known WPW can lead to ventricular fibrillation. In a patient with known WPW with SVT or AF, avoid medications that slow conduction at the AV node such as calcium channel blockers, b-blockers, adenosine, or digoxin. Use procainamide or amiodarone to treat these patients. Infants with WPW syndrome are more likely to have an SVT recurrence and to require antiarrhythmic therapy.66
Summary
Understanding the basic elements of neonatal cardiac anatomy and physiology will help guide the emergency physician when stabilizing critically ill infants prior to transfer to the care of the pediatric cardiologist. Managing an infant with a cardiovascular emergency requires the ability to rapidly assess vital signs, ECG, and CXR at various ages as well as recognition of physical examination clues. Chief complaints can include fussy infants, feeding difficulties, increased work of breathing, wheezing, cyanosis, lethargy, or failure to thrive. While the CHD is a complex group of abnormalities, there are common presentations based on clinically relevant categories that can be used as a framework to approach these patients.
References
1. Silove ED. Assessment and management of congenital heart disease in the newborn by the district pediatrician. Arch Dis Child Fetal Neonatal Ed 1994;70:F71-F74.
2. Steinhorn RH. Evaluation and management of the cyanotic neonate. Clin Ped Emerg Med 2008:9:169-175.
3. Corrall JC. Pediatric heart disease. In: Tintinalli JE, et al, eds. Emergency MedicineA Comprehensive Study Guide, 6th ed. New York: McGraw Hill; 2004:758-769.
4. Yee L. Cardiac emergencies in the first year of life. Emerg Med Clin North Am 2007;25:981-1008.
5. Woolridge DP, Love JC. Congenital heart disease in the pediatric emergency department: Part I: Pathophysiology and clinical characteristics. Pediatr Emerg Med Reports 2002;7:69-81.
6. Savitsky E, Alejos J, Votey S. Emergency department presentations of pediatric congenital heart disease. J Emerg Med 2003;24:239-245.
7. Knowles R, Griebsch I, Dezateux C. Newborn screening for congenital heart defects: A systematic review and cost-effectiveness analysis. Health Technol Assess 2005;9:1-168.
8. Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol 2002;39:1890-1900.
9. Abu-Harb M, Heu E, Wren C. Death in infancy from unrecognized congenital heart disease. Arch Dis Child 1994;71:3-7.
10. Woolridge DP, Love JC. Congenital heart disease in the pediatric emergency department. Part II: Managing acute and chronic complications. Pediatr Emerg Med Reports 2002;7:81-93.
11. Bajolle F, Zaffran S, Bonnet D. Genetics and embryological mechanisms of congenital heart diseases. Arch Cardiovasc Dis 2009;102:59-63.
12. Payne RM, Johnson MC, Grant JW, et al. Toward a molecular understanding of congenital heart disease. Circulation 1995;91:494-504.
13. Allan LD. Fetal cardiology. Curr Opin Obstet Gynecol 1996;8:142-147.
14. Park MK. History Taking. In: Fletcher J, McGonigal C eds. Pediatric Cardiology for Practitioners, 5th ed. Philadelphia: Mosby; 2008:8-26.
15. Onuzo OC. How effectively can clinical examination pick up congenital diseases at birth? Arch Dis Child Fetal Neonatal Ed 2006;91:236-237.
16. Tegnander E, Williams W, Johansen OJ, et al. Prenatal detection of heart defects in a non-selected population of 30,149 fetusesdetection rate and outcome. Ultrasound Obstet Gynecol 2006;27: 252-265.
17. Lee W, Comstock CH. Prenatal diagnosis of congenital heart disease: Where are we now? Ultrasound Clin 2006;1:273-291.
18. Dorfman AT, Marino BS, Wernovsky G, et al. Critical heart disease in the neonate: Presentation and outcome at a tertiary care center. Pediatr Crit Care Med 2008;9:193-202.
19. Wren C, Richmond S, Donaldson L. Presentation of congenital heart disease in infancy: Implications for routine examination. Arch Dis Child Fetal Neonatal Ed 1999;80:49-53.
20. Kim UO, Brousseau DC, Konduri GG. Evaluation and management of the critically ill neonate in the emergency department. Clin Pediatr Emerg Med 2008;9:140-148.
21. Coletti JE, Homme JL, Woodridge DP. Unsuspected neonatal killers in emergency medicine. Emerg Med Clin North Am 2004;22:929-960.
22. Haworth SG, Bull C. Physiology of congenital heart disease. Arch Dis Child 1993;68:707-711.
23. Inaba AS. Cardiac disorders. In: Marx JA, et al, eds. Rosen's Emergency Medicine, 6th ed. Philadelphia: Mosby, 2006:2567-2601
24. Inaba AS. A simple way to remember pediatric vital signs. Contemp Pediatr 2002;1:15.
25. McConnell ME, Adkins SB, Hannon DW. Heart murmurs in pediatric patients: When do you refer? Am Fam Physician 1999; 60:558-565.
26. Sasidharan P. An approach to diagnosis and management of cyanosis and tachypnea in term infants. Pediatr Clin North Am 2004;51:999-1021.
27. Strife JL, Sze RW. Radiographic evaluation of the neonate with congenital heart disease. Radiol Clin North Am 1999;37: 1093-1107.
28. Sharieff GQ, Rao SO. The pediatric ECG. Emerg Med Clin North Am 2006;24: 195-208.
29. Kwon KT, Tsai VW. Metabolic emergencies. Emerg Med Clin North Am 2007;25: 1041-1060.
30. Park MK. Pathophysiology of cyanotic congenital heart defects. In: Fletcher J, McGonigal C, eds. Pediatric Cardiology for Practitioners, 5th ed. Philadelphia: Mosby; 2008:140-168.
31. Gewitz MH, Woolf PK. Cardiac Emergencies. In: Fleisher GR, et al, eds. Textbook of Pediatric Emergency Medicine, 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2006:717-758.
32. Costello JM, Almodovar MC. Emergency care for infants and children with acute cardiac disease. Clin Pediatr Emerg Med 2007;8:145-155.
33. Jones RWA, Baumer JH, Joseph MC, et al. Arterial oxygen tension and response to oxygen breathing in differential diagnosis of congenital heart disease in infancy. Arch Dis Child 1976;51:667-673.
34. Brousseau T, Sharieff GQ. Newborn emergencies: The first 30 days of life. Pediatr Clin North Am 2006;53:69-84.
35. Marino BS, Bird GL, Wernovsky G. Diagnosis and management of the newborn with suspected congenital heart disease. Clin Perinatol 2001;28:91-137.
36. Brown K. The infant with undiagnosed cardiac disease in the emergency department. Clin Pediatr Emerg Med 2005;6:200-206.
37. Penny DJ, Shekerdemian LS. Management of the neonate with symptomatic congenital heart disease. Arch Dis Child Fetal Neonatal Ed 2001;84:141-145.
38. Pickert CB, Moss MM, Fiser DH. Differentiation of systemic infection and congenital obstructive left heart disease in the very young infant. Pediatr Emerg Care 1998;14:263-267.
39. Danford DA, Gutgesell HP, McNamara DG. Application of information theory to decision analysis in potentially prostaglandin-responsive neonates. J Am Coll Cardiol 1986;8:1125-1130.
40. Perry JC, Fenrich AL, Hulse JE, et al. Pediatric use of intravenous amiodarone: Efficacy and safety in critically ill patients from a multicenter protocol. J Am Coll Cardiol 1996;27:1246-1250.
41. Lee C, Custer JW, Rau RE. Drug Doses. In: Custer JW, Rau RE, et al, eds. The Harriet Lane Handbook, 18th ed. Philadelphia: Mosby; 2009:697-1030.
42. Browning KA, Barr P, West M, et al. Transporting newborn infants with suspected duct dependent congenital heart disease on low-dose prostaglandin E1 without routine mechanical ventilation. Arch Dis Child Fetal Neonatal Ed 2007;92: 117-119.
43. Meckler GD, Lowe C. To intubate or not to intubate? Transporting infants on prostaglandin E1. Pediatrics 2009;123: 25-30.
44. Lee C, Mason L. Pediatric cardiac emergencies. Anesthesiol Clin North Am 2001;19:287-308.
45. Gaber KA, McGavin CR, Wells IP. Lateral chest x-ray for physicians. J R Soc Med 2005;98:310-312.
46. Costello JM, Goodman DM, Green TP. A review of the natriuretic hormone system's diagnostic and therapeutic potential in critically ill children. Pediatr Crit Care Med 2006;7:308-318.
47. Johns MC, Stephenson C. Amino-terminal pro-B type natriuretic peptide testing in neonatal and pediatric patients. Am J Cardiol 2008;101:76-81.
48. Ko JK, Deal BJ, Strasburger JF, et al. Supraventricular tachycardia mechanisms and their age distribution in pediatric patients. Am J Cardiol 1992;69: 1028-1032.
49. Ludimorsky A, Garson A. Supraventricular tachycardia. In: Gillette PC, Garson A eds. Pediatric Arrhythmias: Electrophysiology and Pacing. Philadelphia: WB Saunders 1991:380-426.
50. Friedman FD. Intraosseous adenosine for the termination of supraventricular tachycardia in an infant. Ann Emerg Med 1996; 28:356-358.
51. Doniger SJ, Sharieff GQ. Pediatric dysrhythmias. Pediatr Clin North Am 2006;53:85-105.
52. Kugler JD, Danford DA. Management of infants, children, and adolescents with paroxysmal supraventricular tachycardia. J Pediatr 1996;129:324-338.
53. Liberman L. Low energy biphasic waveform cardioversion of atrial arrhythmias in pediatric patients and young adults. Pacing Clin Electrophysiol 2006;29:1383-1386.
54. Lim SH, Anantharaman V, Teo WS, et al. Comparison of treatment of supraventricular tachycardia by valsalva maneuver and carotid sinus massage. Ann Emerg Med 1998;31:30-35.
55. Craig JE, Scholz TA, Vanderhooft SL, et al. Fat necrosis after ice application for supraventricular tachycardia termination. J Pediatr 1998;133:727.
56. Bisset GS, Gaum W, Kaplan S. The ice bag: A new technique for interruption of supraventricular tachycardia. J Pediatr 1980;97:593-595.
57. Atkins DL. Diagnosis and management of supraventricular tachycardia in children. Clin Pediatr Emerg Med 2001;2:107-113.
58. Losek JD, Endom E, Dietrich A, et al. Adenosine and pediatric supraventricular tachycardia in the emergency department: Multicenter study and review. Ann Emerg Med 1999;33:185-191.
59. Getschman SJ, Dietrich AM, Franklin WH. Intraosseous adenosine: As effective as peripheral or central venous administration? Arch Pediatr Adol Med 1994;148:616-619.
60. Ellenbogen KA, Thames MD, DiMarco JP, et al. Electrophysiological effects of adenosine in the transplanted human heart: Evidence of supersensitivity. Circulation 1990;81:821-828.
61. Etheridge SP, Craig JE, Compton SJ. Amiodarone is safe and highly effective therapy for supraventricular tachycardia in infants. Am Heart J 2001;141:105-110.
62. Saul JP, Scott WA, Brown S, et al. Intravenous amiodarone for incessant tachyarrhythmias in children: A randomized, double-blinded, antiarrhythmic drug trial. Circulation 2005;112:3470-3477.
63. Bucknall CA, Keeton BR, Curry PVL, et al. Intravenous and oral amiodarone for arrhythmias in children. Br Heart J 1986; 56:278-284.
64. Kirk CR, Gibbs JL, Thomas R, et al. Cardiovascular collapse after verapamil in supraventricular tachycardia. Arch Dis Child 1987;62:1265-1266.
65. Perry JC, Garson A. Supraventricular tachycardia due to Wolff-Parkinson-White syndrome in children: Early disappearance and late recurrence. J Am Coll Cardiol 1990; 16:1215-1220.
66. Tortoriello TA, Snyder CS, Smith EO. Frequency of recurrence among infants with supraventricular tachycardia and comparison of recurrence rates among those with and without preexitation and among those with and without response to digoxin and/or propranolol therapy. Am J Cardiol 2003;92:1045-1049.
67. Fengler BT, Brady WJ, Plautz CU. Atrial fibrillation in the Wolff-Parkinson-White syndrome: ECG recognition and treatment in the ED. Am J Emerg Med 2007;25: 576-583.
68. González de Dios J, Rodrigues Balo A, Martinez de Azagra Garde, et al. Wolff-Parkinson-White syndrome: Long-term followup study in a pediatric population (85 cases). An Esp Pediatr 1990;32:522-530.
Emergency medicine physicians routinely deal with cardiac emergencies in adult patients but rarely encounter infants with critical cardiac conditions. While the infant's cardiac physiology can be very different from an adult's, the general principles of preload, afterload, contractility, and vascular resistance are the same.Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.