Congenital Heart Disease in the Pediatric Emergency Department: Part II
Part II: Managing Acute and Chronic Complications
Authors: Dale P. Woolridge, MD, PhD, Department of Pediatrics/ Division of Emergency Medicine, Department of Surgery, University of Maryland Medicine, Baltimore; Jon C. Love, MD, Assistant Professor, Division of Pediatric Cardiology, Department of Pediatrics, University of Maryland Medicine, Baltimore.
Peer Reviewer: Perline Ramalanjaona, MD, MA, FAAP, Attending Physician, Department of Pediatrics, Wyckoff Hospital, Brooklyn, NY.
Patients with congenital heart disease (CHD) present a challenge to emergency department (ED) physicians. Presentations vary from an acutely ill child with a new diagnosis of CHD to a child with a known CHD with a routine childhood illness or problems that stem directly from his or her specific heart defect. ED physicians need an understanding of the pathophysiology associated with various CHDs and commonly associated complications. The emergency physician must be able to discriminate between different classes of defects and hence narrow the diagnosis and focus treatment. In addition, the ED physician must be able to recognize common complications that may be associated with certain types of CHD or operative procedures and feel competent in the stabilization and management of these patients until the pediatric intensivist or cardiologist can assume care.
Part I of this two-part series outlined the pathophysiology behind the more common congenital heart defects and discussed the evaluation of these patients. This article focuses on likely presenting conditions and suggested approaches to developing a differential and defining appropriate methods of treatment. In addition, potential co-morbidities in patients who have a congenital heart defect will be discussed.— The Editor
Introduction
Approaching a child with a potential CHD requires a systematic approach. In severe cases, stabilization will be the first priority. Subsequent evaluation entails obtaining clues from history, physical examination, imaging, and laboratory testing. Key diagnostic features may include the presence of cyanosis and its response to oxygen, the quality of peripheral perfusion, precordial auscultation, and palpation. Additional studies should include chest radiography to evaluate pulmonary markings and characteristics of the cardiac silhouette, electrocardiogram (ECG) features, and an upper/lower extremity blood pressure gradient. Age at the time of presentation also can be a key feature in deciding the appropriate treatment for the patient with CHD. The patient who presents acutely in the first week of life is more likely to have a ductal dependent lesion and may require prostaglandins to assist in maintaining ductal patency.1 On the other hand, the child who presents later in infancy with gradually worsening ability to feed and worsening respiratory symptoms is more likely to be in congestive heart failure (CHF) from a left-to-right shunt and require diuresis.
This article completes the process of identification of a child with a new CHD and discusses important management strategies. Conditions in which pre-operative and post-operative patients present to the ED will be explored and guidance offered on the most frequent complications encountered.
Presentation and Treatment
The following section will review the most common presenting scenarios for children with CHD in the ED. The likely underlying cardiac lesions and appropriate treatment strategies will then be discussed. The most common CHDs will be broken down into categories based on the age of the patient at presentation (See Table on Rapid Access Card.). The age when symptoms develop has diagnostic value since this often correlates to a specific phase in the transition from fetal circulation. In the United States, prenatal screening, standardized patient care, and the liberal use of pulse oximetry make it very unlikely that a significant congenital heart defect will remain undiagnosed prior to discharge from the newborn nursery. There are, however, specific cardiac defects that remain elusive in the first few days of life. This is owed primarily to two features characteristic of the newborn: a widely patent ductus arteriosus (PDA) and high pulmonary vascular resistance (PVR). A CHD that presents acutely in the first two weeks of life is much more likely to be a ductal dependent lesion, whereas those children who present later often do so with symptoms of CHF.2 Common CHDs relevant to the ED and their presenting symptoms are summarized in Figure 1. The more severe the defect, the earlier a patient will present.
 |
0-2 Weeks of Life
Congenital heart defects that present to the ED in the first two weeks of life almost exclusively are ductal dependent lesions.3 Stabilization in these patients requires maintaining ductal patency. The mainstay medical therapy for this entails the use of intravenous prostaglandin E1 (PGE1) (See Table on Rapid Access Card.). Prior consultation with a pediatric cardiologist is advisable before starting this drug. Justifiable hesitance to start prostaglandins may be due to an unclear diagnosis, as side effects may antagonize hypotension secondary to sepsis. In addition, there is a relative contraindication to the use of this drug in infants with total anomalous pulmonary venous return (TAPVR). If TAPVR is strongly suspected, it may be prudent to delay the use of prostaglandins until a pediatric cardiology consult is obtained or an echocardiogram is performed. For patients who do have a ductal dependent lesion, intravenous (IV) prostaglandins are life-saving. Therefore, the physician who strongly suspects the presence of an underlying ductal dependent defect in this age group should not hesitate to start prostaglandins. To administer, the dosage of PGE1 is 0.05-0.2 mcg/kg/min as a continuous infusion. Typically, a drip is started at 0.1 mcg/kg/min and gradually may be decreased to 0.05 mcg/kg/min once the patient is stable. Side effects of PGE1 include jitteriness, fever, hypotension, and apnea.1 Therefore, management should include, at a minimum, continuous cardio-respiratory monitoring and a low threshold for intubation, particularly in patients who are to be transferred or remain on prostaglandins for a prolonged period of time.
As mentioned previously, most cyanotic lesions will be diagnosed in the nursery because of the patient’s distinct cyanotic appearance. These patients usually are referred promptly for specialty care and surgical correction. Children with transposition of the great arteries (TGA), hypoplastic left heart syndrome (HLHS), or tetralogy of Fallot (TOF) occasionally may have a delay in diagnosis because they lack distinct cyanosis in the nursery or have a widely patent PDA. These patients will then present with cyanosis (likely in the ED) at a later date. The most common acyanotic lesions that may miss detection and present acutely in the ED are those with critical left heart obstruction such as severe aortic stenosis (AS) and coarctation of the aorta (CoA). These will be discussed in the following sections. CHDs less likely to be seen in the ED will be mentioned briefly.
Transposition of the Great Arteries
TGA is the most common cyanotic CHD that presents in the newborn period. The majority of patients with TGA develop cyanosis within a few hours of birth; only 10% develop cyanosis after the first few days of life. A small minority will have delayed symptoms secondary to a large ventricular septal defect (VSD) or atrial septal defect (ASD), which allows adequate mixing and alleviates some dependence on a PDA. These patients may present as late as 1-2 weeks of life. In addition, these patients often will not have a murmur. Patients typically will present to the ED with complaints pertaining to intermittent cyanosis (exacerbated by crying or feeding) or persistent cyanosis. As the PDA continues to close, hypoxia and acidosis progress. Peripheral pulses will be normal. Other findings include an increased precordial impulse, and a loud/single S2. Chest radiography will reveal the classic "egg on a string" cardiac silhouette in approximately 50% of cases. Respirations are unlabored unless a large VSD is present, which allows for increased pulmonary blood flow. This can be demonstrated by increased pulmonary vascular markings on chest radiography. An ECG is of little diagnostic benefit. One typically sees a sinus tachycardia with right axis deviation.
The initial treatment for these patients should include stabilization and a diagnostic evaluation for sepsis, which is much more common than TGA. The key diagnostic feature that directs the emergency physician toward the possibility of an underlying cardiac defect is the presence of central cyanosis that does not respond to 100% oxygen. Once a diagnosis of TGA is suspected in a patient who is younger than 2 weeks, pediatric cardiology should be consulted promptly and intravenous prostaglandins started if the oxygen saturation is less than 70%.4 The respiratory status should be followed closely. The response to IV prostaglandins is dependent on the presence and size of a coexisting ASD or VSD. For patients who do not respond adequately to PGE1, emergent atrial septostomy is a procedural option available to the cardiologist that often is life-saving.5 This allows for additional left-to-right atrial shunting, distributing oxygenated blood systemically, and alleviating the dependence on a PDA. Additional management may include measures to minimize PVR, which maximizes shunting of deoxygenated blood to the lungs. Therefore, hyperventilation and alkalosis will be beneficial if persistent pulmonary hypertension is present. Blood gases should be checked to monitor the respiratory status and watch for the development of metabolic acidosis secondary to hypoxia. Repeat doses of sodium bicarbonate can be used to correct any acidosis that develops. Ultimately, all medical care is simply palliative until early surgical correction can be performed.
Truncus Arteriosus
Truncus arteriosus (TA) rarely avoids detection in the newborn nursery and, therefore, only will be mentioned briefly here. Physical examination will reveal cyanosis that is directly proportional to the degree of pulmonary blood flow. Patients often will demonstrate bounding pulses secondary to insufficiency of the truncal valve. This insufficiency can be heard as a diastolic murmur greater at the left lower sternal border. Most patients present with progressive symptoms of CHF. Chest radiography shows cardiomegaly, increased pulmonary vascular markings and pulmonary edema. Medical management consists of diuresis and inotropic support. This lesion in itself is not ductal dependent, but prostaglandins may be initiated prior to echocardiography, which characterizes the lesion. Up to 20% of patients with TA may have an associated interrupted aortic arch. These patients are reliant on a patent PDA and will present with signs of cardiogenic shock as the PDA begins to close. In these cases, one will see diminished lower extremity blood pressures and a progressive metabolic acidosis.
Critical Left Heart Obstruction
Critical left heart obstruction (HLHS, AS, CoA, and interrupted aortic arch) is a group of defects that may escape detection in the nursery and subsequently present to the ED. The most common of these is HLHS. Absent or severely diminished left ventricular outflow makes these lesions entirely or partially dependent upon a PDA for systemic perfusion. Typically these patients are symptomatic within the first two days of life, and a diagnosis is made prior to discharge from the nursery. Some patients with delayed PDA closure may continue to appear normal and feed, thus avoiding detection. Patients who are discharged to home eventually will present (as the PDA closes) in cardiogenic shock. The exam reveals marked pallor, poor perfusion, and cyanosis. Additional findings include a single S2 with a hyperdynamic precordium. Pulses are diminished in all extremities. The initial differential must include overwhelming sepsis, and these children should receive a complete septic evaluation and have appropriate antibiotic therapy administered. Children with HLHS may even be febrile due to the existing hypermetabolic state, which may further delay an accurate diagnosis. Cardiogenic shock becomes suspect when hypotension remains refractory to volume resuscitation and/or pressor support. Likewise, central cyanosis may be evident and respond minimally to exogenous oxygen (100% oxygen test). In fact, the administration of oxygen may exacerbate the patient’s condition by further promoting PDA closure or allowing decreased systemic perfusion once the PDA has opened. The chest radiograph often will demonstrate increased pulmonary vascular markings, pulmonary edema, and cardiomegaly. An ECG typically shows a sinus tachycardia with right axis deviation, right atrial enlargement and right ventricular enlargement. It also may reveal signs of myocardial ischemia secondary to poor coronary perfusion.
Pediatric cardiology should be consulted promptly to help guide the care of these patients. Intubation with positive pressure ventilation helps concurrent pulmonary congestion. Ventilator management should be directed toward increasing PVR by targeting a relative hypercarbia (pCO2 of 45-50 mmHg) and hypoxia.6 This promotes improved right-to-left shunting across the PDA for systemic perfusion. Oxygen saturation should be kept well below 90% since (in the one-chamber state) over-perfusion of the lungs occurs at the expense of systemic perfusion. For oxygen saturation approaching 90%, minute ventilation should be reduced. In addition, nitrogen gas can be added to the inspired air to decrease oxygen concentration to below that of room air. It is imperative to balance systemic vascular resistance with PVR. For patients who are hypotensive with poor extremity perfusion, PVR should be increased. Specific treatment requires maintaining ductal patency with continuous intravenous PGE1. Additional management is directed toward reversing the cardiogenic shock. Attempts to improve perfusion pressures with volume or pressors typically are unsuccessful until prostaglandin therapy is started and the PDA opens. Pressors are indicated only if decreased ventricular function is suspected. Acidosis should be corrected and fluid volume deficit replaced. During the process of stabilization, arrangements should be made for transfer or admission and surgical management.
Critical CoA is another lesion that may evade detection in the nursery. Patients may be asymptomatic in the presence of a PDA. Right-to-left flow across the PDA supplies the descending aorta with blood to perfuse systemically when there is preductal obstruction. In addition, obstructions at the level of the PDA often are not restrictive while the PDA remains open. Prior to PDA closure, lower extremity pulses and pressures appear normal. As long as the PDA is widely patent, the patient will remain relatively asymptomatic. Once closure begins, symptoms of shock ensue. Pathognomonic of coarctation is diminished pulses in the lower extremities. Therefore, assessment includes comparisons of pulse oximetry and blood pressure between the right upper extremity and the lower extremity. Chest radiography will demonstrate cardiomegaly, pulmonary edema, and increased vascular markings. An ECG will demonstrate right ventricular hypertrophy in patients who present early (0-2 weeks of life), whereas left ventricular hypertrophy typically develops later.
Maintaining patency of the PDA with IV prostaglandins is life-saving. Additional treatment is directed toward symptoms of shock using fluid replacement, pressor support in the presence of myocardial failure, and correction of any metabolic acidosis. Patients often will present with some degree of ventricular failure requiring inotropic support. If left ventricular failure is present, upper extremity pressures also may be diminished, thus negating the major diagnostic feature of coarctation.7 Diagnosis ultimately is confirmed by echocardiography. In some cases balloon dilation may be palliative until surgical repair is arranged.8
Children with interrupted aortic arch or critical AS often will present similarly to CoA. Symptoms become evident as the PDA closes. An isolated interrupted aortic arch that is proximal to a PDA would demonstrate a difference in upper and lower extremity pulse oximetry similar to that of CoA. Critical AS appears clinically similar to HLHS. Physical findings demonstrate a harsh systolic ejection murmur at the upper sternal border (typically on the right). However, if there is diminished left ventricular function, the characteristic murmur may not be appreciated. Each of these lesions manifests a progressive metabolic acidosis and cardiogenic shock. Stabilization and management is similar to other left heart obstructive defects. IV prostaglandins are essential in maintaining patency of the ductus.
Critical Right Heart Obstruction
Critical right heart obstructive defects rarely avoid detection in the newborn nursery. The exceptions to this are TOF and critical pulmonary stenosis. Both of these lesions will be cyanotic from birth and are highly dependent on the PDA for pulmonary perfusion. Patients will present as the PDA begins to close with a progressive cyanosis. Physical exam will demonstrate tachypnea, tachycardia, and a central cyanosis. Auscultation reveals a loud systolic murmur at the left upper sternal border that may not have been present with a large PDA. Chest radiography demonstrates cardiomegaly (possibly boot-shaped in TOF), and decreased pulmonary vascular markings. Patients with TOF also may have a right-sided aortic arch. When suspected, pediatric cardiology should be consulted and intravenous prostaglandins started if the oxygen saturation is less than 70%. Additional treatment is directed at minimizing PVR to promote left-to-right shunting and pulmonary blood flow. This is done through promoting a respiratory alkalosis through hyperventilation and often requires intubation and mechanical ventilation.
First Year of Life
Congenital heart defects that become symptomatic in the first year of life generally do so with symptoms of CHF.2 These lesions are typically those in which a left-to-right shunt predominates. At birth, shunting is limited by the high PVR in the newborn. Through natural transitioning, PVR drops through the first 6-8 weeks of life and shunting increases proportionately. Initially, a heart murmur often is absent on physical exam and may not be apparent until 2-8 weeks of age when shunting is increased.9 As pulmonary blood flow increases, the patient becomes progressively more tachypneic, resulting in a progressive feeding intolerance, poor weight gain, and irritability. Concomitant sympathetic response results in tachycardia, diaphoresis, and pallor. The development of these symptoms tends to be gradual. Many patients may present with an intercurrent infection as the precipitating factor that pushes the child into symptomatology. Therefore, the evaluating physician should be aware of the potential for underlying cardiac disease when seeing any patient in the ED. Physical findings that may help in the diagnosis include a hyperdynamic precordium and harsh systolic murmur, typically at the left lower sternal border. Tachypnea and hepatomegaly also may be present. Assuming a sizable shunt, evidence of left ventricular hypertrophy is invariably seen on an ECG. Right ventricular hypertrophy is typically a later finding once PVR increases. Treatment is directed toward improving the patient’s respiratory status. Diuresis with furosemide (1-3 mg/kg/dose) usually will provide adequate improvement until the patient can be evaluated by a cardiologist.10 Intubation and positive pressure ventilation may be required for profound signs of CHF. Digoxin typically is not useful in the acute stabilization of the patient and usually is reserved for use by the cardiologist. Long-term management may include digoxin or additional measures to decrease afterload with the use of angiotensin-converting enzyme (ACE) inhibitors.11 Decreasing systemic vascular resistance will help diminish left-to-right shunting. In addition, afterload reduction especially is helpful in the presence of any left ventricular dysfunction.
The most common lesions that present in this fashion are large VSDs, PDAs, and ECDs. Differentiating between these lesions clinically is difficult, and the diagnosis generally is made by echocardiography. All left-to-right shunts secondary to VSDs produce a harsh systolic murmur that is loudest at the left lower sternal border. Bounding pulses also are present due to runoff across the PDA in diastole. ECDs with AV valve insufficiency may have an additional systolic murmur at the apex. ECDs almost always will have extreme left axis deviation (superior axis), whereas the axis is often normal in PDAs and VSDs.
Of all the left-to-right shunt lesions, only the complete ECDs with AV valve insufficiency will present in extremis within the first three months of life. These patients will have symptoms of CHF with chest radiography demonstrating cardiomegaly and pulmonary edema. Next in line of severity are the large VSDs and ECDs with competent AV valves. The majority of these patients are more likely to become symptomatic by 4-6 weeks of life with mild to moderate tachypnea, feeding intolerance, and failure to thrive. Of more importance for these patients is the precipitating event that initiates the patient’s decompensation. These patients have a lower tolerance for routine illnesses, have more respiratory infections, and are more prone to dehydration. Therefore, each patient should be treated on an individual basis.
Another defect that can present later in the first year of life is TOF with mild right heart obstruction. Cyanosis is minimal or transient since the right heart obstruction is mild enough to allow adequate pulmonary blood flow. The clinical course and management of these patients is determined by the associated VSD. Unlike patients who have isolated VSDs, symptoms of CHF and feeding intolerance rarely are appreciated since the right heart obstruction is protective from pulmonary overcirculation. An interesting feature of these patients is intermittent episodes of cyanosis referred to as hypercyanotic spells ("tet spells"). This arises from an acute episode of decreased pulmonary perfusion that is thought to be due to a dynamic component in the right heart obstruction. Medical management for these spells is directed at increasing systemic vascular resistance (similar to the toddler who will stop and squat during these attacks). Other treatments include sedation and IV fluid boluses. The child who is resistant to the above maneuvers should be treated with phenylephrine (5-20 mcg/kg/dose) intravenously.12 Phenylephrine also may be given intramuscularly if necessary.
Infancy to Adolescence
Lesions that present later in infancy or adolescence are left-to-right shunt lesions where shunting is more limited. These include smaller VSDs, partial ECDs, and ASDs. It is of utmost importance that these defects be diagnosed and corrected prior to the development of chronic pulmonary vascular obstructive disease, as this is an irreversible condition. Often these lesions are asymptomatic, with diagnosis relying on the detection of a suspicious murmur. Their presence also may be established following a diagnostic evaluation for stroke or endocarditis. Asymptomatic patients with suspicious murmurs should be referred for evaluation by a pediatric cardiologist. Surgical correction is based on the degree of shunting and risks for the development of chronic pulmonary vascular obstructive disease.
Other lesions that present later include mild CoA and mild AS. CoA most often is diagnosed in infancy by the detection of a suspicious murmur,13 whereas diagnosis in adolescence more often is secondary to hypertension.14 Similarly, patients who are asymptomatic should be referred for outpatient evaluation. For patients symptomatic from hypertension, admission and blood pressure control is warranted. AS often may present in adolescence as an exertional intolerance or syncope. Examination and workup for syncope will reveal left ventricular hypertrophy and a harsh systolic ejection murmur at the right upper sternal border. Often these patients are admitted when echocardiography confirms the diagnosis.
The Preoperative Patient
Due to the many advances in neonatal surgery, most complex congenital heart defects are corrected within the first three months of life. Therefore, the number of infants with CHD who present to the ED who have not undergone corrective or palliative surgery is declining. Despite this, some patient groups awaiting surgery still exist and will require care in the ED. In general, these patients are at risk for sub-optimal weight gain, poor growth, and delayed development. High-calorie formulas, with or without gastric tube placement, help to optimize caloric intake. One preoperative group in particular includes those with left-to-right shunt lesions that have not undergone surgical correction. Others may have had palliative procedures such as balloon dilation in pulmonary stenosis (PS), AS, and mild CoA. These patients often take a diuretic, an afterload reducing agent, or digoxin. Patients who either are not compliant or who have a precipitating illness may present with respiratory decompensation or exacerbation of their CHF. For example, simple gastroenteritis in these patients may lead to significant dehydration and/or inadequate dosing of medications. These patients also are more susceptible to recurrent lower respiratory infections, malnutrition, and bacterial endocarditis. One key feature that often is overlooked is the dosing of medications. As a child grows, the doses of medications should be increased according to the weight gained between clinic visits. It is not an uncommon scenario for a child who presents to the ED to have outgrown the dosage. Often all that is needed is an additional dose of a diuretic and dose adjustments until the patient can follow up with his cardiologist. In this regard, most parents serve as excellent resources when it comes to the patient’s care plan.
Patients with cyanotic heart disease also may present for similar reasons. As mentioned previously, these patients are seen less and less frequently and, for this reason, many physicians are uncomfortable with a patient who regularly sustains an oxygen saturation in the 70s. Although this appears unnatural, one should refrain from excessive administration of oxygen, as this can exacerbate the condition. If adequate tissue oxygenation is in question, one can assess the patient’s acid base status to determine if a metabolic acidosis is developing. In addition, these children will need adequate oxygen-carrying capacity. Therefore, the assessment of a hemoglobin and hematocrit level may be beneficial in the symptomatic patient. Patients with uncorrected cyanotic CHD also are at particular risk for failure to thrive and delayed development. These patients require close follow up for tight medical management. For this reason, it is essential that the managing cardiologist be aware of any concerns that arise in the ED.
One problem that may need addressing in the TOF patient who hasn’t undergone surgical correction is hypercyanotic or "tet" spells. TOF patients who have mild right heart obstruction may undergo repair at a later age. Since obstruction is mild, pulmonary blood flow is adequate, and patients often are managed medically on an outpatient basis prior to surgical repair. These hypercyanotic or "tet" spells are episodes in which the patient has an acute increase in right heart obstruction resulting in diminished pulmonary blood flow. Management of these patients basically involves increasing systemic vascular resistance to overcome the increased right heart obstruction. Initially this may involve bringing the knees to the chest to simulate the classic "squatting" seen in older patients who have not undergone surgical repair. Other treatments include sedation and IV fluid boluses. The child who is resistant to the above maneuvers should be treated with phenylephrine (5-20 mcg/kg/dose) intravenously.12 Phenylephrine also may be given intramuscularly if necessary.
Another group of patients that should be mentioned here is the group in which correction is being performed in a staged process. B-T shunts can be placed palliatively in cases of cyanotic CHD. (See Table 1.) This shunt is at risk of obstruction and/or thrombosis manifesting as a progressive cyanosis. In these cases, systemic pressures should be augmented with volume and/or pressor support to ensure optimal flow across the shunt. Emergent echocardiography, possibly followed by catheterization, is required to assess the status of the shunt. B-T shunts also may allow too much flow to be conducted to the pulmonary vasculature. The earliest sign of this is higher than expected oxygen saturations: i.e., oxygen saturations consistently in the upper 80s to 90s. Over time, patients may develop symptoms of CHF. Chest radiography may reveal increased pulmonary vascular markings and pulmonary edema. Patients would, therefore, benefit from diuresis and, if CHF is severe, intubation with positive pressure ventilation. Staged repair also occurs with HLHS. These patients invariably are followed closely by tertiary care centers. Presenting conditions can include symptoms secondary to output failure or CHF. A specific cause of decompensation in these patients stems from recurrent aortic arch obstruction following the staged Norwood (first stage) correction. It is important to evaluate these patients for evidence of this when they present to the ED. Contact should be made with the managing cardiologist to direct patient care.
 |
Another condition that patients with unrepaired CHD are at risk for is stroke. CHD is the most common cause of stroke in the pediatric patient population.15 Fortunately, stroke is a rare occurrence in pediatrics. Any patient with cyanotic disease (due to the implied presence of right-to-left shunting) are at highest risk but patients with left-to-right shunt lesions also are at risk. Those who present acutely with a sudden onset of neurologic deficit and who fulfill criteria should be considered candidates for thrombolytic therapy.15
Routine Peri-operative Care and Operative Repair
Left-to-right Shunt Lesions. In all left-to-right shunt lesions, increased pressure and volume are transmitted to the pulmonary vasculature. Over time, the resultant pulmonary hypertension will lead to the development of right heart failure and irreversible chronic pulmonary vascular obstructive disease. The rate in which this develops is directly proportional to the degree of shunting and, hence, the degree of pulmonary hypertension.
In ASDs and VSDs, the decision for operative repair is, for the reasons above stated, made on an individual basis.16 The goal of therapy is to promote appropriate growth and development and to avoid the complications of pulmonary vascular disease. Medical management for these lesions is directed at preventing signs and symptoms of CHF. This entails the use of furosemide 1-3 mg/kg/day to improve pulmonary congestion, digoxin and/or an ACE inhibitor to decrease systemic pressures and limit left-to-right shunting. Traditionally, indication for surgical closure of ASDs is a pulmonary to systemic blood flow ratio of 1.5:1 as measured by cardiac catheterization. However, these patients are no longer taken for cardiac catheterization since the development of improved echocardiography. In current practice, if the defect is large enough to produce right heart enlargement on echocardiography it should be considered for surgical closure.17 For the emergency physician unfamiliar with echocardiography, ECG changes suggestive of right atrial enlargement may serve an equivalent indicator. For large lesions that do not close or are not expected to close, recommended treatment is surgical closure at 2-4 years of age.18 For patients with VSDs who are clinically asymptomatic but with evidence of pulmonary hypertension, correction should be performed by 1 year of age.19 Lesions in older patients with normal pulmonary artery pressures should be repaired if pulmonary to systemic blood flow is greater than 2:1.
PDAs rarely are symptomatic in the full-term infant. Pre-term infants are much more likely to be symptomatic and require earlier intervention. Indomethacin is effective in initiating closure in many pre-term infants.20 Term infants, on the other hand, tend to be unresponsive to indomethacin and, therefore, require either coil occlusion through cardiac catheterization or surgical ligation.21 For patients who present with CHF and have failed attempted closure with indomethacin, treatment requires ligation. PDAs are the only CHD that can be considered "cured" after ligation.
Physiologically, ECDs act in a similar manner to VSDs. Therefore, the goal of therapy is directed at avoiding symptoms of CHF and the development of chronic pulmonary vascular obstructive disease. Patients who have recurrent respiratory infections or poor weight gain will be repaired earlier. Incomplete ECDs require a repair tailored to the individual lesion. The inlet type of defect tends to not close spontaneously and, if it is sufficiently large, is often repaired at earlier ages.22
Critical CoA is corrected emergently in patients with cardiomegaly, signs of circulatory shock, severe hypertension, and severe CHF.23 Catheter-mediated balloon dilation may be palliative in these cases but often does not replace surgical correction. Asymptomatic patients with mild stenosis should undergo elective repair, which may include balloon dilation through cardiac catheterization, between 3 and 10 years of age.24 Re-stenosis can occur in up to 16% of cases and often can be managed with repeat balloon dilation25 or stent placement26 within the narrowing. In cases where surgery is delayed, medical management is directed at controlling blood pressure and diuresis to avoid the development of CHF.
The treatment of AS varies and is based on the degree of obstruction with corrective options, including surgical valvotomy.25 Here, the native pulmonary valve is removed and replaced with a conduit or homograft from the right ventricle to the pulmonary artery. The pulmonary valve is then used as the aortic valve. Of importance is the need to re-implant the coronary vessels. Unfortunately, not all patients are candidates for a Ross procedure. These patients may require prosthetic valves that carry the added risks associated with anticoagulation therapy. This can give rise to complications and will be discussed later. Another option for these patients is percutaneous balloon valvuloplasty.27 This often is used as a first-line therapy to allow the patient to grow before surgery is required. Balloon dilation has been shown to be associated with re-stenosis and may require repeat dilation.
HLHS is a ductal dependent lesion that is incompatible with life. Treatment consists of initial stabilization. Therapy is directed at maintaining patency of the ductus arteriosus using intravenous prostaglandins and maintaining elevated PVR to promote right-to-left shunting. Mechanical ventilation may assist in maintaining an increased PVR.28 Ventilator settings are used to limit pO2 and elevate pCO2 through decreasing inhaled oxygen and decreasing minute ventilation, respectively. Each of these contribute to an increase in PVR. Once stabilized, surgical correction can be considered. This typically involves a three-step process. In the first stage, the hypoplastic aorta is reconstructed using the pulmonary artery to maintain adequate systemic perfusion, with the right ventricle used as the systemic ventricle.29 In addition, a shunt is formed from the aorta to the branch pulmonary arteries that allows restricted flow. This is accomplished with the first step of the Norwood procedure and results in both pulmonary and systemic venous return mixing at the level of the morphologic right ventricle. The first stage usually is performed between 3 days to 3 weeks of life depending on the age of presentation.30 The second step usually is performed between 4 and 6 months of age and entails a hemi-Fontan procedure. This involves removing the aortic to pulmonary artery shunt (placed during the first stage) and connecting the superior vena cava directly into the pulmonary artery. The third and final step completes the Fontan procedure, or total caval pulmonary anastomosis. This is done at approximately 1 year of age and serves to direct blood flow from the inferior vena cava to the pulmonary artery. After the final stage, the systemic venous and pulmonary venous blood flow will be separated, thus mimicking normal cardiovascular circulation. Therefore, low PVR is crucial to the success of the second and third steps. These patients are, therefore, more susceptible to pulmonary disease as well as dehydration. Another option at some pediatric cardiovascular centers is cardiac transplantation.31 The limitations of this option are based on finding donors of the right size and match.
TOF can have varied degrees of presentation. If the right heart obstruction is severe, the lesion is ductal dependent and, therefore, incompatible with life. Definitive treatment for these patients is surgical correction. This is done by patch closure of the VSD and surgical relief of the right heart outlet obstruction. Today, surgery is performed during the first year of life with most occurring within the first three months. Palliative surgery is rarely performed due to the improved outcomes with the complete repair during the newborn period. If a complete repair is not possible, an aortic to pulmonary artery shunt is performed to improve pulmonary blood flow until a complete repair can be achieved.32
Tricuspid valve atresia, owing to the resultant hypoplasia of the right ventricle, requires completion of a Fontan repair as described by the last two stages for HLHS. These patients do not require aortic reconstruction as with patients with HLHS, but may need a palliative arterial shunt (Blalock-Taussig shunt) during the newborn period if there is limited pulmonary blood flow.
Pulmonary valve atresia with a VSD is palliated during the newborn period with an arterial shunt, allowing closure of the PDA. Eventually they will undergo surgical correction with patch closure of the VSD and placement of a conduit (or homograft) between the right ventricular outflow and the pulmonary artery (Rastelli repair). Correction of pulmonary valve atresia without a VSD has a spectrum of options that primarily depend on the size of the right ventricle and tricuspid valve. Many can be staged to a four-chamber repair as with pulmonary atresia/VSD or undergo a Fontan repair. One very important feature, which can be associated with pulmonary valve atresia without a VSD, is the possibility of abnormal coronary artery connections with the right ventricle.
Patients with TGA are highly reliant on a PDA. Therefore, it is necessary that this remains patent and may be done medically with the use of intravenous prostaglandin. If there continues to be significant cyanosis despite prostaglandins, there often is severe restriction of the atrial communication that must be relieved by balloon atrial septostomy.5,33 The resultant large ASD will allow adequate left-to-right atrial shunting and alleviate the need for a PDA. Once stable, surgical repair by the arterial switch procedure typically is performed within 7-14 days or earlier in most centers.34 Repair beyond this time frame is associated with an increased risk of left ventricular failure at the time of the arterial switch.35
Medical management of TA is palliative until surgical correction can be performed. These patients are at high risk for the development of chronic pulmonary vascular disease. Medical therapy with diuresis and digoxin is, therefore, directed at minimizing pulmonary over circulation. ACE inhibitors are used to decrease systemic vascular resistance.11 Surgical repair is done by using the truncus as the new aorta and creating a communication from the right ventricle to the pulmonary arteries, which are detached from the common trunk.36 Patch closure of the VSD is performed concurrently. As the child grows, the conduit will need to be replaced with a larger one to accommodate increasing blood flow. Placement of a new conduit is typically performed at 3-6 years of age and again in adolescence.37 Up to 20% of these defects also may be associated with an interrupted aortic arch. For these children, PGE1 may be essential for maintaining lower extremity perfusion until surgical repair can be performed.
Patients with TAPVR tend to have significant pulmonary vascular congestion and often present immediately after birth with respiratory distress. Positive pressure ventilation helps to diminish pulmonary congestion. For these patients, emergent ultrasound is used to characterize the anomalous vessels. Operative repair then can be performed to reconstruct pulmonary venous return to the left atrium.38 Patients with a more delayed presentation are those who have only partial TAPVR, often with a sinus venosus ASD. These patients are not cyanotic but have severe right heart enlargement. Patients with milder defects should be managed medically for several months to improve operative outcomes.38 Medical management for TAPVR is directed toward decreasing pulmonary blood flow with the use of diuretics and digoxin. The use of PGE1 in emergent cases remains controversial.
The Postoperative Patient
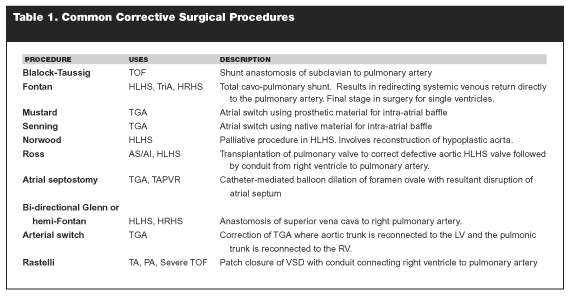
Patients who have undergone operative repair of congenital heart defects are at increased risk of developing complications stemming from the original defect and the procedure used to correct that defect. These potential conditions are specific to the surgical techniques used for correction. Common surgical procedures and a brief description are listed in Table 1. Improved techniques have allowed for an increasing population of long-term survivors. In addition, patients who underwent outdated surgical procedures that are now abandoned are presenting later in life with complications. For this reason, it is helpful for the emergency physician to have a basic knowledge of the more common corrective procedures and the constellation of side effects that may be associated with them.
The Mustard and Senning procedures are referred to collectively as atrial switch operations. These were performed from 1964-1985 for the correction of TGA. The procedure entailed constructing an atrial baffle using prosthetic material (Mustard) or native tissue (Senning) that directed systemic venous blood to the left atrium and pulmonary venous blood to the right atrium. The right ventricle remained the systemic ventricle and the left ventricle remained the pulmonary ventricle. As the patients grew older, it was noted that the right ventricle often was inadequate at maintaining systemic demands and eventually failed.39 In addition, there was a significant rate of sudden death in these patients with the primary risk factor being development of atrial dysrhythmia.40 For this reason, these techniques were abandoned in the mid-80s and replaced by the arterial switch operation.41 Typically, failure following the atrial switch procedure is not seen until the second postoperative decade. Late complications of the Mustard procedure were noted by Puley et al.42 These include: dysrhythmias (66%), CHF (10%), and sudden death (9%). Of the dysrhythmias noted, 35% were atrial flutter, and 48% had at least one episode of supraventricular tachycardia. Twenty-two percent of these patients required pacemaker placement.
Complications that are associated with surgical conduits include stenosis and endocarditis. Pulmonary atresia (PA) with or without a VSD is the typical patient who may undergo surgery involving the placement of a conduit (Rastelli procedure). Other heart defects where correction entails the placement of a conduit also include: TA, PA, and TriA. All patients will outgrow their conduits over time, requiring replacement as they become more restrictive to the increased demands of a larger heart. Replacement usually is required at approximately 5-6 years of age and in adolescence. Obstruction may develop gradually or acutely from a thrombovascular event.43 A common area of obstruction in patients who underwent TA repair with a conduit is at the branch pulmonary arteries. Symptoms of conduit obstruction are hepatomegaly, peripheral edema, and jugular venous distention (superior vena cava syndrome). Hypotension and cardiovascular collapse is a late finding and may be associated with acute thrombosis of the conduit. Other shunts that are at risk of obstruction include the Blalock-Taussig (B-T) shunt and the Glenn shunt (hemi-Fontan procedure) which connect the pulmonary artery to the subclavian vein or superior vena cava, respectively.44 The Blalock-Taussig shunt is used as a palliative treatment for CHD with decreased pulmonary blood flow. Ideally, this shunt should have a mild degree of obstruction to protect the pulmonary vasculature from over-perfusion. Patients who are obstructing this shunt often present with worsening cyanosis and a loud murmur. The Glenn shunt is used in single ventricle conditions such as HLHS and TriA. Obstruction of a bi-directional Glenn shunt would most likely present with signs of SVC syndrome. Patients who present acutely in any of these conditions and are known to have specific shunts should be considered candidates for heparinization and thrombolytics. It is imperative that the managing cardiologist be made aware to assist in management decisions and further evaluation.
Another complication that merits discussion here is that of myocardial ischemia. Both the Ross and Arterial switch procedures entail coronary artery re-implantation. This puts patients at increased risk for short-term and long-term thrombo-occlusion at these sites. Otherwise, the risk of myocardial ischemia in patients who underwent surgical correction of CHD is owed primarily to their underlying disease. Risk factors include the presence of hypertrophy, myocarditis, cardiomyopathy, CHF, and high output demands. The evaluation of these patients will invariably require that an ECG be obtained. Many patients who have undergone surgical corrections will have abnormalities on the ECG that are without clinical significance. It is, therefore, wise to obtain a previous ECG for comparison to determine whether there are changes that suggest ischemia.
Dysrhythmias
Many congenital heart defects predispose the patient to the development of dysrhythmias. The treatment of these rhythms is in itself an extensive topic and will, therefore, not be discussed here. Three primary factors that initiate abnormal rhythms include: congenital abnormalities in the conduction system that are associated with the structural defect; abnormal conduction pathways or properties that arise from surgical instrumentation or scar formation; and irritable foci in diseased myocardium as a result of the abnormal hemodynamics. Surgical predispositions typically are derived from the region of the conduction system instrumented. Impingement on the sinoatrial node and atrioventricular (AV) node can result in sick sinus syndrome and AV block, respectively.45 Surgical incision also causes an area of conduction block along the incision site that may serve as an axis for reentrant tachycardias. Most surgeons will use an atrial approach if access to the internal structures of the heart is needed. Interestingly, TOF repair is the one of the few defects that often requires a ventriculotomy. This combined with a predisposition to dysrythmia is thought to contribute to the high rate of sudden death seen on late follow-up.46 Atrial reconstruction with atriotomy also will strongly predispose to reentry tachydysryhthmias.47 Some congenital heart defects are associated with specific dysrhythmias. For example, Wolf-Parkinson-White (WPW) syndrome is associated with Ebstein’s anomaly of the tricuspid valve and corrected L-TGA. AV block is associated with repair of TGA, VSDs, ASDs, and ECDs and may be permanent in 1-2% of patients postoperatively.48 Permanent AV block is the most common indication for pacemaker placement in the pediatric population.49
The development of dysrhythmias in patients with congenital heart defects is highest in the early postoperative period. Most of these will spontaneously resolve. For those dysrhythmias that cannot be controlled or suppressed with medical management, catheter ablation often is successful.50 Placement of a defibrillator and/or pacer is another option for the electrophysiologist.49
The most common dysrhythmia on long-term follow-up of patients with CHD is atrial flutter. Atrial fibrillation, and sick sinus syndrome also are frequently seen.51 Rarer are reentry rhythms, AV block, and ventricular dysrhythmias. Factors that contribute to the development of these rhythms include age, underlying cardiac dysfunction, and atrial hypertrophy.52 Atrial flutter may occur in 35% of TOF children who undergo repair, and 25% who have the Fontan or Mustard procedure on long-term follow-up.40,53,54 Flutter episodes in these patients often are sustained and require direct cardioversion.55 Amiodarone as a preventative antiarrhythmic is being used more frequently for the control of supraventricular tachycardia in the pediatric population.56 Ventricular dysrhythmias are relatively rare but pose a significant health concern. Factors that contribute to the development include a history of ventriculotomy (scar mediated), underlying atrial tachycardia, underlying cardiac dysfunction, and proarrhythmia effect of antiarrhythmic medications. A published population based study of sudden death in patients with CHD demonstrating tachycardia at the time of resuscitation was highest following AS repair (13%), followed by patients who had undergone Mustard or Senning procedure (9%), TOF (2%), and CoA (1%) repairs.56
Summary
CHD can be a very complex topic. This article attempts to give the emergency physician an adequate understanding of the pathophysiology involved so accurate decisions can be made while evaluating these patients. Presenting conditions can be quite varied, and, based on the underlying defect, require different levels of care. The patient with complex disease who is managed medically does not tolerate exacerbations well. Undiagnosed patients can present at any time requiring emergent life-sustaining treatment. Poor decisions can have fatal outcomes. Congenital heart defects and the associated surgical corrections have with them a host of complications and additional risks. These patients are at high risk for the development of arrhythmia, endocarditis (See Insert), respiratory infections, and even stroke. Both early and late postoperative complications mandate a keen awareness to the potential complications that are associated with each corrective procedure.
References
1. Reddy SC, Saxena A. Prostaglandin E1. First stage palliation in neonates with congenital cardiac defects. Indian J Pediatr 1998;65:211-216.
2. Burton DA, Cabalka AK. Cardiac evaluation of infants. The first year of life. Pediatr Clin N Am 1994;41:991-1015.
3. Flynn PA, Engle MA, Ehlers KH. Cardiac issues in the pediatric emergency department. Pediatr Clin N Am 1992;39:955-968.
4. Lees MH, King DH. Cardiogenic shock in the neonate. Pediatr Rev 1988;9: 258-266.
5. Baylen BG, Grzeszczak MB, Gleason ME, et al. Role of balloon atrial septostomy before early arterial switch repair of transposition of the great arteries. J Am Coll Cardiol 1992;19:1025.
6. Atz AM, Feinstein JA, Jonas RA. Preoperative management of pulmonary venous hypertension in hypoplastic left heart syndrome with restrictive atrial septal defect. Am J Cardiol 1999;83:224-228.
7. Demircin M, Arsan S, Pasaoglu I, et al. Coarctation of the aorta in infants and neonates: Results and assessment of prognostic variables. J Cardiovasc Surg 1995;36:459-464.
8. Fawzy ME, Sivanandam V, Galal O, et al. One- to ten-year follow-up results of balloon angioplasty of native coarctation of the aorta in adolescents and adults. J Am Coll Cardiol 1997;30:1542-1546.
9. Pelech AN. The cardiac murmur. Pediatr Clin N Am 1998;45:107-122.
10. Driscoll DJ. Left to right shunt lesions. Pediatr Clin N Am 1999;46:355-368.
11. Calligaro IL, Burman CA. Pharmacologic considerations in the neonate with congenital heart disease. Clin Perinatol 2001;28:209-222.
12. Evans-Berro EA. How to defeat a "tet spell." Am J Nurs 1991;91:46-48.
13. Ing FF, Starc TJ, Griffiths SP, et al. Early diagnosis of coarctation of the aorta in children: A continuing dilemma. Pediatrics 1996;98:378-382.
14. Brouwer R, Erasmus ME, Ebels T, et al. Influence of age on survival, late hypertension and recoarctation in elective aortic coarctation repair. J Thorac Cardiovasc Surg 1994;108:525-532.
15. DeVeber G, Roach ES, Riela AR, et al. Stroke in children: Recognition, treatment, and future directions. Semin Pediatr Neurol 2000;7:309-317.
16. Kidd L, Driscoll D, Gersony W, et al. Second Natural History Study of Congenital Heart Defects: Results of treatment of patients with ventricular septal defects. Circulation1993;87:38-51.
17. Friedli B, Oberhansli I, Faidutti B. Interventional catheterization in surgically treated patients with congenital heart disease. Thorac Cardiovasc Surg 2000;48:319-322.
18. Murphy JG, Gersh BJ, McGoon MD, et al. Long-term outcome after surgical repair of isolated atrial septal defect. Follow-up at 27 to 32 years. N Eng J Med 1990;13:323:1645-1650.
19. Niwa K, Perloff JK, Kaplan S, et al. Eisenmenger syndrome in adults: Ventricular septal defect, truncus arteriosus, univentricular heart. J Am Coll Cardiol 1999;34:223-232.
20. Barst RJ, Gersony WM. The pharmacological treatment of patent arteriosus. A review of the evidence. Drugs 1989;38:249-266.
21. Shanthala CC, Maiya PP, Vishwanath D, et al. Clinical profile and management of PDA in neonates. Indian J Pediatr 1997;64:667-670.
22. Brickner M, Hillis LD, Lange RA. Congenital heart disease in adults. New Engl J Med 2000;342:256-262.
23. Van Son JAM, Falk V, Schneider P, et al. Repair of coarctation of the aorta in neonates and young infants. J Cardiol 1997;12:139-146.
24. Pihkala J, Nykanen D, Freedom RM. Interventional cardiac catheterization. Pediatr Clin N Am 1999;46:441-464.
25. Zehr KJ, Gillinov AM, Redmond JM, et al. Repair of coarctation of the aorta in neonates and infants: A thirty-year experience. Ann Thorac Surg 1995;59: 33-41.
26. Hamdan MA, Maheshwari S, Fahey JT, et al. Endovascular stents for coarctation of the aorta: Initial results and intermediate-term follow-up. J Am Coll Cardiol 2001;38;1518-1523.
27. Vahanian A. Balloon valvuloplasty. Heart 2001;85:223-228.
28. Fedderly RT. Left ventricular outflow obstruction. Pediatr Clin North Am 1999;46:369-384.
29. Bove EL, Lloyd TR. Staged reconstruction for hypoplastic left heart syndrome. Ann Surg 1996;224:387-395.
30. Bove EL. Current status of staged reconstruction for hypoplastic left heart syndrome. Pediatr Cardiol 1998;19:308-315.
31. Razzouk Al, Chinnock RE, Gundry SR, et al. Transplantation as a primary treatment for hypoplastic left heart syndrome: Intermediate-term results. Ann Thorac Surg 1996;62:1-8.
32. Waldman JD, Wernly JA. Cyanotic congenital heart disease with decreased pulmonary blood flow in children. Pediatr Clin N Am 1999;46:385-404.
33. Shrivastava S. Timing of surgery/catheter intervention in common congenital cardiac defects. Indian J Pediatr 2000;67:2-6.
34. Soongswang J, Adatia I, Newman C, et al. Mortality in potential arterial switch candidates with transposition of the great arteries. J Am Coll Cardiol 1998;32:753-757.
35. Aseervatham R, Pohlner P. A clinical comparison of arterial and atrial repairs for transposition of the great arteries: Early and midterm survival and functional results. Aust N Z J Surg 1998;68:206-208.
36. Imamura M, Drummond-Webb JJ, Sarris GE, et al. Improving early and intermediate results of truncus arteriosus repair: A new technique of truncal valve repair. Ann Thorac Surg 1999;67:1142-1146.
37. McElhinney DB, Rajasinghe HA, Mora BN, et al, Reinterventions after repair of common arterial trunk in neonates and young infants. J Am Coll Cardiol 2000;35:1317-1322.
38. Harris MA, Valmorida JN. Neonates with congenital heart disease, Part IV: Total anomalous pulmonary venous return. Neonatal Netw 1997;16:63-66.
39. Helbing W, Hansen B, Ottenkamp J, et al. Long-term results of atrial correction for transposition of the great arteries. J Thorac Cardiovasc Surg 1994; 108:363-372.
40. Gelatt M, Hamilton RM, McCrindleBW, et al. Arrhythmia and mortality after the mustard procedure: A 30-year single center experience. Pediatr Cardiol 1997;29:194-201.
41. Planche C, Lacour-Gayet F, Serraf A. Arterial switch. Pediatr Cardiol 1998; 19:297-307.
42. Puley G, Siu S, Connelly M, et al. Arrhythmia and survival in patients >18 years of age after the mustard procedure for complete transposition of the great arteries. Am J Cardiol 1999;83:1080-1084.
43. Kreutzer J. Transcatheter intervention in the neonate with congenital heart disease. Clin Perinatol 2001;28:137-157.
44. Marx GR, Allen HD, Ovitt TW, et al. Balloon dilation angioplasty of Blalock-Taussig shunts. Am J Cardiol 1998;62:824-827.
45. Bharati S, Lev M. Sequelae of atriotomy and ventriculotomy on the endocardium, conduction system and coronary arteries. Am J Cardiol 1982;50: 580-587.
46. Saul JP, Alexander ME. Preventing sudden death after repair of tetralogy of Fallot: Complex therapy for complex patients. J Cardiovasc Electrophysiol 1999;10:1271-1287.
47. Paul T, Windhagen-Mahnert B, Kriebel T. Atrial reentrant tachycardia after surgery for congenital heart disease: Endocardial mapping and radiofrequency catheter ablation using a novel, noncontact mapping system. Circulation 2001;103:2266-2271.
48. Weindling SN, Saul JP, Gamble WJ, et al. Duration of complete atrioventricular block after congenital heart disease surgery. Am J Cardiol 1998;82: 525-527.
49. Gregoratos G, Cheitlin MD, Conill A, et al. ACC/AHA guidelines for implantation of cardiac pacemakers and antiarrhythmia devices: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 1998;31:1175-1209.
50. Freidli B, Oberhansli I, Faidutti B. Interventional catheterization in surgically treated patients with congenital heart disease. Thorac Cardiovasc Surg 2000;48:319-322.
51. LeRoy SS. Clinical dysrhythmias after surgical repair of congenital heart disease. AACN 2001;12:87-99.
52. Fishberger SB, Wernovsky G, Gentles TL, et al. Factors that influence the development of atrial flutter after the Fontan operation. J Thor Cardiovasc Surg 1997;113:80-86.
53. Driscoll DJ, Offord KP, Feldt RH, et al. Five- to fifteen-year follow-up after Fontan operation. Circulation 1992;85:469-496.
54. Haines DE, DiMarco JP. Sustained intra-atrial reentrant tachycardia: Clinical, electrocardiographic and electrophysiologic characteristics and long-term follow-up. J Am Coll Cardiol 1990;15:1345-1354.
55. Etheridge SP, Craig JE, Compton SJ. Amiodarone is safe and highly effective therapy for supraventricular tachycardia in infants. Am Heart J 2001;141: 105-110.
56. Silka MJ, Hardy BG, Menashe VD, et al. A population-based prospective evaluation of risk of sudden cardiac death after operation for common congenital heart defects. J Am Coll Cardiol 1998;32:245-251.
Subscribe Now for Access
You have reached your article limit for the month. We hope you found our articles both enjoyable and insightful. For information on new subscriptions, product trials, alternative billing arrangements or group and site discounts please call 800-688-2421. We look forward to having you as a long-term member of the Relias Media community.